

Key Points
ARIEL functions as an enhancer RNA to mediate the chromatin-chromatin interaction between the ARID5B enhancer and promoter in T-ALL cells.
ARIEL plays an oncogenic role by activating the oncogenic transcriptional program in TAL1+ T-ALL cells.

Abstract
The oncogenic transcription factor TAL1 regulates the transcriptional program in T-ALL. ARID5B is one of the critical downstream targets of TAL1, which further activates the oncogenic regulatory circuit in T-ALL cells. Here, we elucidated the molecular functions of the noncoding RNA, ARID5B-inducing enhancer associated long noncoding RNA (ARIEL), in T-ALL pathogenesis. We demonstrated that ARIEL is specifically activated in TAL1+ T-ALL cases, and its expression is associated with ARID5B enhancer activity. ARIEL recruits mediator proteins to the ARID5B enhancer, promotes enhancer-promoter interactions, and activates the expression of ARID5B, thereby positively regulating the TAL1-induced transcriptional program and the MYC oncogene. The TAL1 complex coordinately regulates the expression of ARIEL. Knockdown of ARIEL inhibits cell growth and survival of T-ALL cells in culture and blocks disease progression in a murine xenograft model. Our results indicate that ARIEL plays an oncogenic role as an enhancer RNA in T-ALL.
Introduction
T-cell acute lymphoblastic leukemia (T-ALL) is a hematological malignancy characterized by clonal proliferation of T-cell precursors arising from the thymus.1-3 One of the most frequent genetic abnormalities is the dysregulation of transcription factor genes, including the overexpression of TAL1 and activating mutations in NOTCH1.4,5 TAL1 is a class II basic helix-loop-helix (bHLH) family transcription factor. It is normally expressed in hematopoietic stem cells (HSCs) and progenitor cells, and is downregulated during T-cell development.6 TAL1 is abnormally expressed in T-ALL cases due to chromosomal abnormalities or somatic mutations in an enhancer element.7-9 TAL1 regulates gene expression by forming a complex that includes the class I bHLH E-proteins (E2A and HEB), GATA family proteins, and LMO family proteins.6 RUNX1, MYB, and the ETS family proteins often co-occupy the regulatory elements with TAL1 and coordinately regulate the expression of target genes.7,10-12
Our group has previously identified several protein-coding genes that are directly activated by TAL1 in T-ALL cells, including AT-rich interactive domain-containing protein 5B (ARID5B).10,13-15 ARID5B functions as a transcriptional activator that can positively regulate the expression of TAL1, its regulatory partners (GATA3, RUNX1, and MYB), and the MYC oncogene, thereby regulating the oncogenic transcriptional program in T-ALL.13 ARID5B is a potential pro-oncogenic factor that supports the survival of human T-ALL cells in culture, and its forced expression in immature T cells results in thymus retention, radioresistance, and tumor formation in zebrafish. Notably, ARID5B is normally downregulated during the double-negative 2 to 4 stages of thymocytes, but the ARID5B −135-kb enhancer bound by the TAL1 complex is activated in T-ALL cells but not in normal thymocytes or mature CD4+ T cells. This locus is associated with a superenhancer that exhibits high levels of surrogate markers of active enhancers, such as histone H3K27ac.16-18 However, the detailed mechanisms underlying the activation of the ARID5B enhancer remain unclear.
Recent genome-wide studies have demonstrated that >80% of the human genome is actively being transcribed.19-22 Long noncoding RNAs (lncRNAs) have been reported to be important for cellular homeostasis, and the dysregulation of lncRNAs can contribute to diseases, such as cancer.23-26 Certain types of noncoding RNAs termed “enhancer RNAs (eRNAs)”27,28 or “ncRNA-activating RNAs (ncRNA-a)” have been reported.29 The eRNAs are actively transcribed from enhancer regions30 and are essential for sustaining enhancer activities. They are involved in the regulation of gene expression through various mechanisms, including RNA polymerase II (Pol II) recruitment to the promoter, stabilization of enhancer-promoter looping, and recruitment of methyltransferase complexes to open chromatin sites.30,31 Several lncRNAs have also been implicated in T-ALL.32-34 The LUNAR1 is directly regulated by NOTCH1 and activates the expression of the IGF1 gene as an eRNA.32 Additionally, we recently established a bioinformatics pipeline that identified 2236 putative lncRNA loci expressed in TAL1+ T-ALL cells.34 By integrating this result with our previously generated chromatin immunoprecipitation (ChIP) sequencing (ChIP-seq) data sets, we selected lncRNAs that were transcriptionally activated by TAL1 under superenhancers in T-ALL cells but not in normal thymocytes. This list included a novel lncRNA (XLOC_005968) that was expressed at the ARID5B −135-kb enhancer locus. Notably, this lncRNA overlapped with the last 3 exons of the CABCOCO1/C10ORF107 gene; however, the full-length CABCOCO1 gene was not expressed in the T-ALL cells. This lncRNA was specifically expressed in several TAL1+ T-ALL cell lines and primary T-ALL cases in our cohort.
In this study, we elucidated the molecular functions and oncogenic roles of XLOC_005968 associated with the ARID5B enhancer in T-ALL. This lncRNA functions as an eRNA to mediate the chromatin-chromatin interaction between the ARID5B enhancer and promoter and plays an important role in the TAL1-induced transcriptional program.
Materials and methods
Cell culture
All leukemia and lymphoma cell lines were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (BioWest). The 293T cells were cultured in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (BioWest). The cell viability was measured using CellTiter-Glo (Promega).
Gene knockdown and CRISPR/CAS9
The short-hairpin RNA (shRNA) were cloned into the lentivirus vector pLKO.1-puro. Single-guide RNAs (sgRNA) were cloned into the lentivirus vector pLV-hU6-sgRNA-hUbC-dCAS9-KRAB-T2a_puro (gifted from Charles Gersbach [Addgene #71236]). The individual shRNA or sgRNA construct was cotransfected into 293T cells with the packaging plasmids pMDLg/pRRE and pRSV-Rev and the envelope plasmid VSV-G using the FuGENE 6 reagent (Roche). Supernatants containing the lentivirus were collected and infected into T-ALL cells in the presence of polybrene (Sigma-Aldrich). The shRNA and sgRNA sequences were shown in supplemental Table 1 (available on the Blood Web site).
Immunoblotting
The cell pellets were lysed in radioimmunoprecipitation assay buffer with protease inhibitor cocktail (Roche). Equal amounts of protein from each sample were diluted in Laemmli sample buffer containing β-mercaptoethanol (Bio-Rad), resolved on an sodium dodecyl sulfate polyacrylamide gel electrophoresis, and transferred onto a polyvinylidene difluoride membrane (Bio-Rad). The membrane was blocked with nonfat milk, probed with the designated primary and secondary antibodies, and subsequently developed using the SuperSignal West chemiluminescence reagent (Thermo Fisher). Antibody information is provided in supplemental Materials and methods.
qRT-PCR and ddPCR
The total RNA was extracted using RNeasy Mini kit (QIAGEN). Human thymus RNA was purchased from Clontech. RNA was reverse-transcribed using QuantiTect (QIAGEN) and RNA expression levels were determined by quantitative reverse transcription polymerase chain reaction (qRT-PCR) using a QuantStudio 3 Real-Time PCR system (Thermo Fisher Scientific) and Power SYBR Green PCR Master Mix (Roche). The digital droplet PCR (ddPCR) experiment was performed using a QX200 Droplet Digital PCR system (Bio-Rad). The PCR was prepared using QX200 ddPCR EvaGreen Supermix (Bio-Rad) and Droplet generation oil for EvaGreen (Bio-Rad Laboratories). The data were processed using QuantaSoft (v.1.7.4). The primer sequences are listed in supplemental Table 2.
RNA-seq
The total RNA was extracted using the miRNeasy kit (Qiagen). The RNA samples were treated with the Turbo DNA-free kit (Ambion). The strand-specific library construction and sequencing of the paired-end 100-bp–long reads using Illumina HiSeq 4000 were performed at BGI Biotech. The RNA-sequencing (RNA-seq) data were analyzed as previously described13 and detailed methodologies can be found in supplemental Material and methods.
Chromatin immunoprecipitation
The chromatin immunoprecipitation (ChIP) assay was performed as previously described.10 Detailed methodologies including antibody information is provided in supplemental Material and methods. The ChIP-PCR primer sequences used are listed in supplemental Table 3. All ChIP-seq data were processed and aligned to the human genome sequence (hg19) as previously described.13
Circular chromatin conformation capture sequencing
The cells were first cross-linked with formaldehyde. The extracted DNA was digested with the primary restriction enzyme HindIII-HF (New England Biolabs) and was subjected to proximity ligation using T4 DNA ligase (Thermo Fisher Scientific), followed by reverse cross-linking. A second restriction enzyme digestion using DpnII (New England Biolabs) was then conducted, followed by a circularization reaction using T4 DNA ligase. Inverse nested PCR was conducted and the products were run using Tris/Borate/EDTA–polyacrylamide gel electrophoresis. DNA purified from the gels was applied for direct sequencing using an Illumina Miseq.
RNA immunoprecipitation
The cells were first cross-linked in formaldehyde. After being flash-frozen, the pellets were lysed with radioimmunoprecipitation assay lysis buffer and sonicated using a Bioruptor sonicator. The supernatant was collected and mixed with an equal amount of the native lysis buffer. The lysate was incubated with an antibody, followed by the addition of protein G Dynabeads. After washing and the reverse-crosslinking, the RNA was extracted using a RNeasy Min-elute column (Qiagen).
RNA pulldown
The sense or antisense strand of ARIEL was cloned into the pCS2 expression plasmid, which was used for in vitro transcription using the MEGAscript T7/SP6 Transcription kit (Ambion). The transcribed RNA was tagged with biotin and purified using the Pierce RNA 3′ End Desthiobiotinylation kit (Thermo Fisher Scientific). The biotinylated RNA was incubated with the precleared cell lysate and then Dynabeads MyOne Streptavidin C1 (Life Technologies). The bound protein was subsequently eluted in Laemmli buffer.
Chromatin isolation by RNA purification
The cells were cross-linked with glutaraldehyde. After the addition of lysis buffer, the cells were sonicated. The chromatin was isolated and probed with the antisense oligos against ARIEL or negative control. Dynabeads MyOne Streptavidin C1 were added for isolation followed by the reverse cross-linking. The probe sequences are listed in supplemental Table 4.
Mouse xenograft
Mouse studies strictly adhered to the recommendations of the institutional animal care and use committee, and all protocols were approved by the Committee at the National University of Singapore. MOLT-4 cells expressing the luciferase gene were transduced with an shRNA. After selection, the equal number of cells were injected into the NSG mice (InVivos). Luminescence signals were detected using the IVIS Spectrum In Vivo Imaging System (PerkinElmer).
Statistical analysis
Significant values (P values) were calculated using 2-tailed Student t tests. The Fisher exact test was conducted using hypergeometric distribution.35 All P values ≤.05 were considered statistically significant.
Dataset availability
Gene expression data sets for 2 cohorts of primary T-ALL samples reported by the TARGET project36 and by Wallaert et al33 were downloaded from the dbGAP database (phs000464) and GEO (GSE89978), respectively. Accession numbers for ChIP-seq data sets for transcription factor proteins and H3K27ac have been previously reported and can be found in supplemental Table 5.
Results
The expression of XLOC_005968/ARIEL is associated with the activity of the ARID5B enhancer
We first investigated the activity of the ARID5B enhancer and its effect on the expression of a novel lncRNA (XLOC_005968) located at this locus. By ChIP-seq analysis, we observed high levels of H3K27ac signal at the −135-kb element in TAL1+ T-ALL cell lines (Jurkat and MOLT-4) but not in normal T cells (thymus, Th1, Th2, and Th17) (Figure 1A). The H3K4me1 signal, which represents an enhancer element, and mediator binding were also observed at the same element (Figure 1A red arrowhead). In contrast, the H3K4me3 signal, which represents a promoter element, was much higher around the transcriptional start site of the ARID5B gene (black arrowhead). In an independent ChIP-PCR analysis, H3K4me1 was confirmed to be more enriched around the −135-kb element than H3K4me3, whereas H3K4me3 was more enriched at the ARID5B promoter site (supplemental Figure 1A). Importantly, the −135-kb locus was bound by the TAL1 complex in ChIP-seq (Figure 1A), followed by an independent validation by ChIP-PCR (supplemental Figure 1B-C). The luciferase assay using a reporter construct encoding this sequence also confirmed that the −135-kb enhancer activity was positively regulated by each member of the TAL1 complex (supplemental Figure 1D-E).
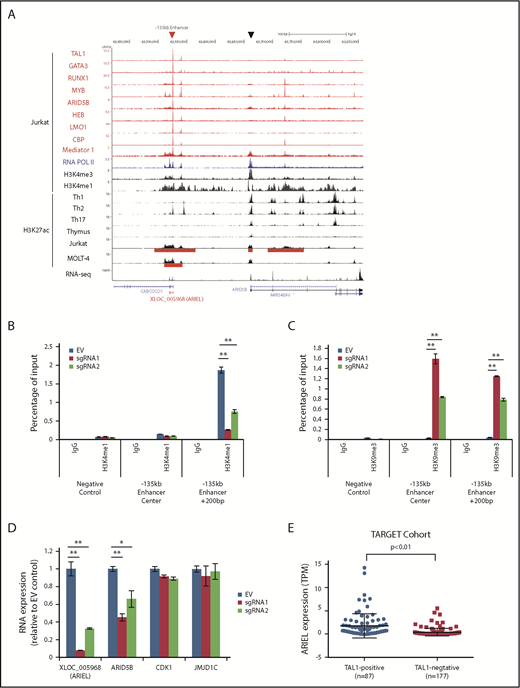
XLOC_005968/ARIEL expression is associated with the activity of the ARID5B −135-kb enhancer. (A) ChIP-seq gene tracks displaying DNA binding of TAL1, and its regulatory partners, CBP, mediator 1 and RNA Pol II in Jurkat cells as well as various histone markers in CD4+ T cells (Th1, Th2, and Th17), normal thymus, Jurkat, and MOLT-4 cells at the XLOC_005968/ARIEL locus. RNA-seq data (positive strand) in Jurkat is shown. Superenhancer regions defined by the H3K27ac ChIP-seq analysis are indicated (red bars). A red arrowhead and a black arrowhead indicate the ARID5B −135-kb enhancer and promoter, respectively. (B-C) Jurkat cells were cotransduced with the catalytic-dead form of the dCas9-KRAB protein and an sgRNA (sgRNA1 or sgRNA2) or an empty vector (EV). ChIP analysis was performed using a specific antibody against H3K4me1 (B), H3K9me3 (C), or control IgG. The amount of genomic DNA in the ChIP and input samples was measured by qPCR using specific primers targeting the ARID5B −135-kb enhancer locus or a region +200 bp downstream from the center. The IGFBP3 promoter region was used as a negative control for ChIP-PCR. The percentage of the input is shown as the mean plus or minus standard deviation (SD) of triplicate experiments. (D) The RNA expression levels of XLOC_005968/ARIEL, ARID5B, and neighboring genes (CDK1 and JMJD1C) were analyzed by qRT-PCR. The expression was normalized to the ERCC RNA spike-in and is shown as the following values relative to the EV control: mean plus or minus SD of triplicate experiments. (E) Expression of ARIEL from the RNA-Seq data set reported by Liu et al36 was first calculated and reported as transcripts per million (TPM). Expression of ARIEL between TAL1+ and TAL1− patients were then analyzed using nonpaired Student t test. **P < .01.
XLOC_005968/ARIEL expression is associated with the activity of the ARID5B −135-kb enhancer. (A) ChIP-seq gene tracks displaying DNA binding of TAL1, and its regulatory partners, CBP, mediator 1 and RNA Pol II in Jurkat cells as well as various histone markers in CD4+ T cells (Th1, Th2, and Th17), normal thymus, Jurkat, and MOLT-4 cells at the XLOC_005968/ARIEL locus. RNA-seq data (positive strand) in Jurkat is shown. Superenhancer regions defined by the H3K27ac ChIP-seq analysis are indicated (red bars). A red arrowhead and a black arrowhead indicate the ARID5B −135-kb enhancer and promoter, respectively. (B-C) Jurkat cells were cotransduced with the catalytic-dead form of the dCas9-KRAB protein and an sgRNA (sgRNA1 or sgRNA2) or an empty vector (EV). ChIP analysis was performed using a specific antibody against H3K4me1 (B), H3K9me3 (C), or control IgG. The amount of genomic DNA in the ChIP and input samples was measured by qPCR using specific primers targeting the ARID5B −135-kb enhancer locus or a region +200 bp downstream from the center. The IGFBP3 promoter region was used as a negative control for ChIP-PCR. The percentage of the input is shown as the mean plus or minus standard deviation (SD) of triplicate experiments. (D) The RNA expression levels of XLOC_005968/ARIEL, ARID5B, and neighboring genes (CDK1 and JMJD1C) were analyzed by qRT-PCR. The expression was normalized to the ERCC RNA spike-in and is shown as the following values relative to the EV control: mean plus or minus SD of triplicate experiments. (E) Expression of ARIEL from the RNA-Seq data set reported by Liu et al36 was first calculated and reported as transcripts per million (TPM). Expression of ARIEL between TAL1+ and TAL1− patients were then analyzed using nonpaired Student t test. **P < .01.
To examine the effect of ARID5B enhancer activity on the lncRNA expression, we next designed 2 independent sgRNAs that bind the center of the −135-kb element and delivered each sgRNA together with a FLAG-tagged dead Cas9 (dCas9) fused with a transcriptional repressor peptide (KRAB).37 This system can physically block and epigenetically silence the target region. The binding of FLAG-dCas9-KRAB was confirmed by ChIP-PCR analysis (supplemental Figure 1F). In this setting, we observed a significant decrease in the H3K4me1 levels at +200 bp of the −135-kb element after the sgRNA transduction (Figure 1B). This change was accompanied by an increase in the H3K9me3 levels at the same element (Figure 1C), which is a repressive histone marker. Importantly, we observed significant decreases in the expression of both XLOC_005968 and ARID5B but not of other neighboring genes (CDK1 and JMJD1C) (Figure 1D; supplemental Figure 1G). Additionally, knockdown of each member of the TAL1 complex downregulated XLOC_005968 expression (supplemental Figure 1H-I), confirming the finding in our previous study.34 These results indicated that the expression of XLOC_005968 is associated with the ARID5B enhancer activity. Hence, we here termed this lncRNA “ARID5B inducing enhancer-associated long noncoding RNA (ARIEL)”.
We then analyzed the expression of ARIEL in 2 other cohorts of primary T-ALL samples. Analysis of gene expression of the TARGET data set with 264 primary T-ALL samples36 showed that ARIEL were significantly highly expressed in TAL1+ cases compared with TAL1− T-ALL patients (Figure 1E). We were also able to observe a modest positive correlation between ARIEL and ARID5B expression among the TAL1+ patients who express ARIEL (supplemental Figure 1J). We also analyzed a microarray data set for 64 primary T-ALL samples reported by Wallaert et al33 and the expression of ARIEL (detected by probe A_32_P8156) was significantly higher in the TAL1+ cases than in the other T-ALL subgroups (supplemental Figure 1K). These results further supported that ARIEL is specifically expressed in TAL1+ T-ALL.
ARIEL positively regulates ARID5B expression in T-ALL cells
We next analyzed potential molecular functions of ARIEL. We first performed subcellular fractionation, followed by a digital droplet qRT-PCR analysis, to analyze the localization of the ARIEL in the T-ALL cells. The purity of the fractions was verified by western blot (supplemental Figure 2A). This analysis demonstrated that ARIEL was present both in the cytoplasmic and nuclear fractions in Jurkat and MOLT-4 cells (Figure 2A-B).

ARIEL positively regulates ARID5B expression in T-ALL cells. (A-B) The number of copies of ARIEL transcripts in each subcellular fraction was measured by performing digital droplet qPCR in Jurkat (A) and MOLT-4 (B) cells and shown as arbitrary unit (A.U.). (C-D) Jurkat (C) and MOLT-4 (D) cells were transduced by lentivirus infection with either a control shRNA (shGFP) or an shRNA targeting ARIEL (sh3 and sh5). RNA expression of ARIEL, ARID5B, and neighboring genes (CDK1 and JMJD1C) was measured by qRT-PCR 3 days after the transduction and normalized to GAPDH expression. Expression values relative to the control shGFP sample are shown as the mean plus or minus SD of triplicate experiments. (E) The protein expression of ARID5B in the Jurkat and MOLT-4 cells was analyzed by western blot analysis after the shRNA transduction. α-tubulin was used as an internal control. (F) Jurkat cells were transduced with either a control shRNA (shGFP) or an shRNA targeting ARID5B (sh3 and sh7). See panel C legend for details.
ARIEL positively regulates ARID5B expression in T-ALL cells. (A-B) The number of copies of ARIEL transcripts in each subcellular fraction was measured by performing digital droplet qPCR in Jurkat (A) and MOLT-4 (B) cells and shown as arbitrary unit (A.U.). (C-D) Jurkat (C) and MOLT-4 (D) cells were transduced by lentivirus infection with either a control shRNA (shGFP) or an shRNA targeting ARIEL (sh3 and sh5). RNA expression of ARIEL, ARID5B, and neighboring genes (CDK1 and JMJD1C) was measured by qRT-PCR 3 days after the transduction and normalized to GAPDH expression. Expression values relative to the control shGFP sample are shown as the mean plus or minus SD of triplicate experiments. (E) The protein expression of ARID5B in the Jurkat and MOLT-4 cells was analyzed by western blot analysis after the shRNA transduction. α-tubulin was used as an internal control. (F) Jurkat cells were transduced with either a control shRNA (shGFP) or an shRNA targeting ARID5B (sh3 and sh7). See panel C legend for details.
Because ARIEL can be localized in the nucleus and its expression is associated with ARID5B enhancer activity, we hypothesized that ARIEL might serve as an eRNA to regulate the expression of neighboring genes. To analyze the effect of ARIEL expression on gene regulation, we designed 2 shRNAs specifically targeting the ARIEL transcript. We were able to efficiently knock down ARIEL as confirmed by qRT-PCR in Jurkat and MOLT-4 cells (Figure 2C-D). Importantly, we observed a significant decrease in the messenger RNA (mRNA) expression of ARID5B after knockdown of ARIEL, whereas other actively transcribed neighboring genes (CDK1 and JMJD1C) were hardly affected. This change was accompanied by a decrease in the protein levels of ARID5B in both cell lines (Figure 2E). Similar results were observed in 2 other TAL1/ARIEL+ T-ALL cell lines, CTV-1 and HSB-2 (supplemental Figure 2B-E). Importantly, we did not find any changes in ARID5B expression levels upon transduction of ARIEL shRNA in 2 TAL1+ T-ALL cells (CCRF-CEM and RPMI 8402) that do not express ARIEL (supplemental Figure 2B,F-G). Thus, the effects observed on ARID5B expression in ARIEL+ cells are likely due to specific depletion of ARIEL and not to off-target effects of the shRNAs.
Notably, the knockdown of ARID5B also resulted in a decrease in the expression of ARIEL without affecting the expression of other neighboring genes (Figure 2F). The ChIP-seq and luciferase reporter assay showed that ARID5B protein can bind at its own enhancer element (Figure 1A) and positively regulate its activity (supplemental Figure 1D-E). These results indicated that ARIEL and ARID5B positively regulate each other, thus forming a positive feed-forward loop.
ARIEL affects the TAL1-induced regulatory program by promoting ARID5B expression
To analyze the genes and pathways affected by ARIEL in T-ALL, we then performed an RNA-seq analysis after knocking down of ARIEL in Jurkat cells. Among the top 100 genes that were significantly downregulated after the knockdown (adjusted P < .05, log2 fold change less than −0.6), ARID5B was selected (supplemental Figure 3A). The Gene Set Enrichment Analysis (GSEA) demonstrated that ARID5B target genes tended to be also downregulated following knockdown of ARIEL (P < .001; Figure 3A), suggesting that the loss of ARIEL affects the gene expression program regulated by ARID5B. It is noted that although only 25 of the top downregulated genes was commonly selected between ARID5B and ARIEL knockdown by regular statistical analysis using cutoff values (adjusted P < .05, log2 fold change less than −0.6), the overlap was significant by the Fisher exact test (P < 5.5 × 10−14) (supplemental Figure 3B) and thus was not simply due to chance. One possibility for the small number of overlapping genes is due to the difference of time course and ARID5B expression level after each knockdown because the effect of ARIEL on these target genes are indirect (secondary) through ARID5B. It also suggests that ARIEL might have effects independent of affecting ARID5B and the TAL1 complex.
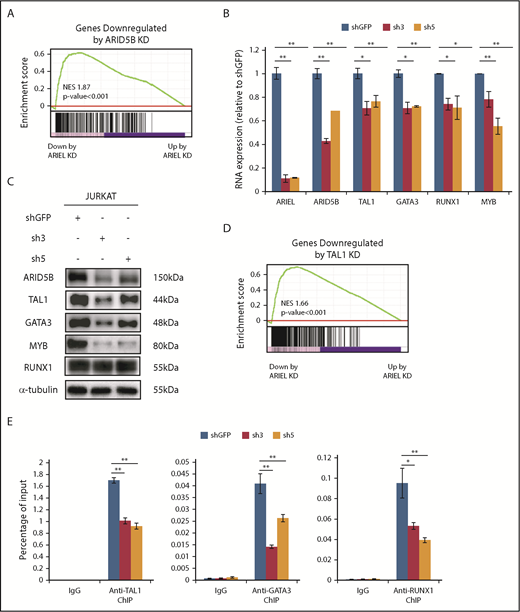
ARIEL affects the TAL1-induced regulatory program through the regulation of ARID5B expression. (A) The high-confidence ARID5B target genes that were bound by ARID5B protein and downregulated by ARID5B knockdown in Jurkat cells were previously determined13 and used as a gene set. GSEA was performed to analyze the overall correlation of the gene expression changes following the ARIEL knockdown in the same cells. Normalized enrichment score (NES) and P values are shown. (B) The RNA expression of ARID5B and each member of the TAL1 complex was measured by qRT-PCR after the shRNA knockdown of ARIEL in Jurkat cells. See Figure 2C legend for details. (C) Western blot analysis showing the protein expression of each member of the TAL1 complex in the Jurkat cells after the ARIEL knockdown. (D) The high-confidence TAL1 target genes bound by TAL1 and downregulated after the TAL1 knockdown in the Jurkat cells were used as a gene set. See panel A legend for details. (E) ChIP analysis was performed using a specific antibody against TAL1, GATA3, RUNX1, or control IgG in Jurkat cells after the knockdown of ARIEL using shRNAs (sh3 and sh5). The amount of genomic DNA in the ChIP and input samples was measured by qRT-PCR using specific primers targeting the ARID5B −135-kb enhancer locus. The percentage of the input is shown as the mean plus or minus SD of duplicate experiments.
ARIEL affects the TAL1-induced regulatory program through the regulation of ARID5B expression. (A) The high-confidence ARID5B target genes that were bound by ARID5B protein and downregulated by ARID5B knockdown in Jurkat cells were previously determined13 and used as a gene set. GSEA was performed to analyze the overall correlation of the gene expression changes following the ARIEL knockdown in the same cells. Normalized enrichment score (NES) and P values are shown. (B) The RNA expression of ARID5B and each member of the TAL1 complex was measured by qRT-PCR after the shRNA knockdown of ARIEL in Jurkat cells. See Figure 2C legend for details. (C) Western blot analysis showing the protein expression of each member of the TAL1 complex in the Jurkat cells after the ARIEL knockdown. (D) The high-confidence TAL1 target genes bound by TAL1 and downregulated after the TAL1 knockdown in the Jurkat cells were used as a gene set. See panel A legend for details. (E) ChIP analysis was performed using a specific antibody against TAL1, GATA3, RUNX1, or control IgG in Jurkat cells after the knockdown of ARIEL using shRNAs (sh3 and sh5). The amount of genomic DNA in the ChIP and input samples was measured by qRT-PCR using specific primers targeting the ARID5B −135-kb enhancer locus. The percentage of the input is shown as the mean plus or minus SD of duplicate experiments.
Because ARID5B can directly regulate the expression of TAL1 and its regulatory partners,13 we validated the expression of these genes and their downstream targets after the ARIEL knockdown using 2 independent shRNAs in multiple cell lines. Strikingly, the expression levels of TAL1, GATA3, RUNX1, and MYB were significantly decreased at the mRNA level (Figure 3B) in the Jurkat cells following the knockdown of ARIEL. Protein expression levels were also decreased, except RUNX1 protein, which were hardly affected possibly due to the protein stability (Figure 3C). Similar results were observed in the MOLT-4 cells (supplemental Figure 3C-D). The GSEA using several gene sets that defined the target genes of each transcription factor revealed that most downstream target genes were downregulated following the knockdown of ARIEL (Figure 3D; supplemental Figure 3E-G). Furthermore, based on the ChIP-PCR analysis, TAL1, GATA3, and RUNX1 DNA binding at the ARID5B −135-kb enhancer was significantly reduced after the knockdown of ARIEL (Figure 3E), suggesting that the downregulation of the TAL1 complex members further affects ARID5B enhancer activity. These observations indicated that ARIEL can affect the TAL1-induced transcriptional program by regulating ARID5B.
ARIEL recruits the mediator complex to the ARID5B −135-kb enhancer
Subsequently, we sought to identify the molecular mechanism by which ARIEL regulates the ARID5B expression. Because several eRNAs have been known to regulate chromatin-chromatin interactions though interacting with mediator proteins,29,32 we first examined whether the loss of ARIEL affects the binding of mediator proteins at the ARID5B regulatory elements. Strikingly, we observed a significant decrease in the binding of the mediator 1 (Figure 4A) and mediator 12 (Figure 4B) proteins at the −135-kb enhancer element but not at the ARID5B promoter after the knockdown of ARIEL in the Jurkat cells. Furthermore, the decreased binding of the mediator complex was accompanied by the reduced binding of RNA Pol II at both the −135-kb enhancer and ARID5B promoter regions (Figure 4C). Similar results were observed in the MOLT-4 cells (supplemental Figure 4A-C).
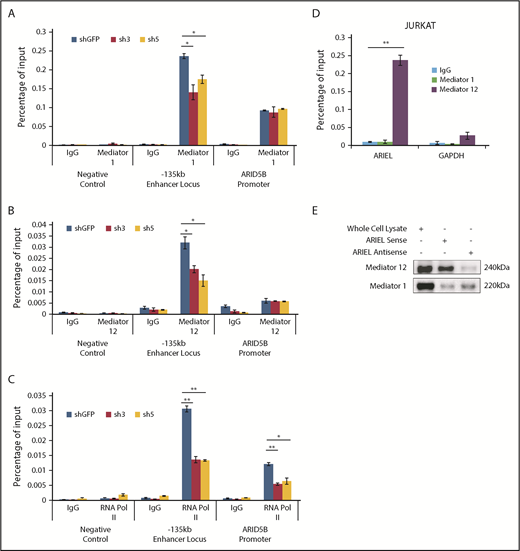
ARIEL recruits the mediator complex to the ARID5B −135-kb enhancer locus. (A-C) ChIP analysis was performed using a specific antibody against mediator 1 (A), mediator 12 (B), or RNA POL II (C) or control IgG in Jurkat cells after the shRNA knockdown of ARIEL. See Figure 1B legend for details. (D) Jurkat cells were subjected to a RIP assay using a specific antibody against mediator 1 or mediator 12 or control IgG. The RNA transcripts in the RIP lysate and input samples were measured by qRT-PCR using specific primers targeting ARIEL or GAPDH (negative control). The percentage of the input is shown as the mean plus or minus SD of triplicate experiments. (E) The sense or antisense strand of ARIEL was biotinylated and mixed with the whole-cell lysate from Jurkat cells. The in vitro pulldown assay was conducted and western blot analysis was performed to detect the presence of the mediator 12 protein in the pulldown lysate.
ARIEL recruits the mediator complex to the ARID5B −135-kb enhancer locus. (A-C) ChIP analysis was performed using a specific antibody against mediator 1 (A), mediator 12 (B), or RNA POL II (C) or control IgG in Jurkat cells after the shRNA knockdown of ARIEL. See Figure 1B legend for details. (D) Jurkat cells were subjected to a RIP assay using a specific antibody against mediator 1 or mediator 12 or control IgG. The RNA transcripts in the RIP lysate and input samples were measured by qRT-PCR using specific primers targeting ARIEL or GAPDH (negative control). The percentage of the input is shown as the mean plus or minus SD of triplicate experiments. (E) The sense or antisense strand of ARIEL was biotinylated and mixed with the whole-cell lysate from Jurkat cells. The in vitro pulldown assay was conducted and western blot analysis was performed to detect the presence of the mediator 12 protein in the pulldown lysate.
Then, we analyzed the physical interaction between ARIEL RNA and the mediator complex. We performed an RNA immunoprecipitation (RIP) assay using specific antibodies against the mediator 1 and 12 proteins in Jurkat cells, followed by qRT-PCR analysis. We verified that those proteins were efficiently pulled down (supplemental Figure 4D-E). Importantly, compared with a negative control transcript (GAPDH), we detected high amounts of ARIEL transcripts in RIP eluates of mediator 12 in both the Jurkat and MOLT-4 cells (Figure 4D; supplemental Figure 4F). We did not detect significant amounts of ARIEL in the RIP eluates of mediator 1, suggesting that ARIEL might interact primarily with the mediator 12 protein.
We also conducted reciprocal in vitro pulldown experiments using a biotinylated ARIEL transcript incubated in total cell lysate harvested from Jurkat cells. Similar to the observations in the RIP assay, we detected enrichment of the mediator 12 protein but very minimal mediator 1 after the pulldown with the sense ARIEL transcript (Figure 4E). The antisense ARIEL RNA could pull down the mediator 12 protein to a much lesser extent, suggesting that the interaction between ARIEL and the mediator 12 protein is sequence specific and is not due to promiscuous binding.
ARIEL promotes the ARID5B enhancer-promoter interaction
We next performed a 4C-seq analysis to investigate whether the interaction between the ARID5B enhancer and promoter can be affected by ARIEL expression. We designed 1 PCR primer near the −135-kb element (“viewpoint”) and another primer near the ARID5B promoter region. In this analysis, we observed strong interactions between these regions in the control sample as shown by the heatmap (Figure 5A top, red arrowhead). This interaction was nearly entirely lost following the knockdown of ARIEL (bottom). Notably, active enhancer markers, such as H3K4me1 and H3K27ac, at the −135-kb element or the ARID5B promoter region were not significantly affected after ARIEL knockdown (supplemental Figure 5A-B). Thus, ARIEL may not be involved in the activation or maintenance of the histone status.
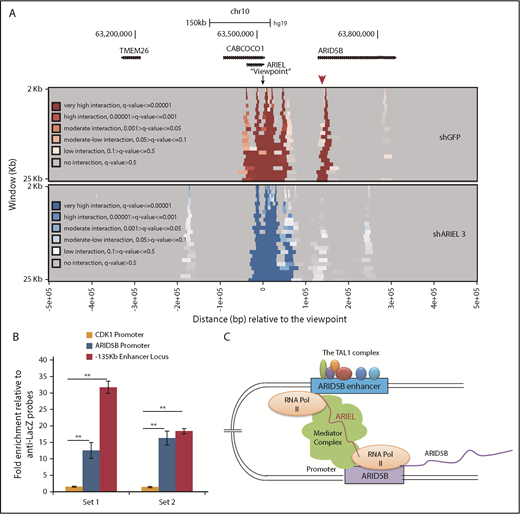
ARIEL promotes the ARID5B enhancer-promoter interaction. (A) Jurkat cells were transduced with either a control shRNA (shGFP) or an shRNA targeting ARIEL (sh3) by lentivirus infection. Genomic DNA samples were harvested on day 3 of the shRNA transduction and subjected to a 4C assay. A heatmap showing the interactions from the viewpoint (ARID5B −135-kb enhancer locus) of the neighboring genomic regions before and after the knockdown of ARIEL. Color codes represent the quantitative scale (q-value). A red arrowhead indicates the ARID5B promoter. (B) ChIRP assay was performed using 2 different sets of antisense probes (“set 1” and “set 2”) that detect ARIEL or control probes (anti-Lacz). The amount of genomic DNA in the ChIP and input samples was measured by performing qPCR using specific primers targeting the ARID5B −135-kb enhancer locus or the ARID5B promoter region. The neighboring locus (CDK1 promoter) was used as a negative control region. The fold-enrichments are shown as the mean plus or minus SD of duplicate experiments. (C) Proposed model.
ARIEL promotes the ARID5B enhancer-promoter interaction. (A) Jurkat cells were transduced with either a control shRNA (shGFP) or an shRNA targeting ARIEL (sh3) by lentivirus infection. Genomic DNA samples were harvested on day 3 of the shRNA transduction and subjected to a 4C assay. A heatmap showing the interactions from the viewpoint (ARID5B −135-kb enhancer locus) of the neighboring genomic regions before and after the knockdown of ARIEL. Color codes represent the quantitative scale (q-value). A red arrowhead indicates the ARID5B promoter. (B) ChIRP assay was performed using 2 different sets of antisense probes (“set 1” and “set 2”) that detect ARIEL or control probes (anti-Lacz). The amount of genomic DNA in the ChIP and input samples was measured by performing qPCR using specific primers targeting the ARID5B −135-kb enhancer locus or the ARID5B promoter region. The neighboring locus (CDK1 promoter) was used as a negative control region. The fold-enrichments are shown as the mean plus or minus SD of duplicate experiments. (C) Proposed model.
Finally, we examined whether ARIEL can recognize the ARID5B enhancer or promoter region. We performed chromatin isolation using an RNA purification (ChIRP) assay to specifically pull down genomic regions. First, we confirmed that the 2 sets of biotinylated antisense probes (“set 1” and “set 2”) could specifically pull down the ARIEL transcript but not the negative control (GAPDH) (supplemental Figure 5C). Then, we analyzed the enrichment of the specific genomic DNA elements that were pulled down with each probe. Strikingly, we observed significant enrichments in the DNA amount of the −135-kb enhancer and promoter regions after the pulldown using both antisense probes compared with that in a negative control region (CDK1 promoter) (Figure 5B). Thus, ARIEL can localize in the ARID5B enhancer and promoter regions in the nucleus. Our results are consistent with the model that ARIEL can facilitate the ARID5B enhancer-promoter interaction by recruiting the mediator complex to specific genomic elements (Figure 5C).
ARIEL is required for T-ALL cell growth and survival in culture
Next, we investigated the roles of ARIEL in T-ALL cell maintenance. We first analyzed cell growth and survival after ARIEL knockdown in Jurkat and MOLT-4 cells. Interestingly, we observed a significant decrease in cell growth in both cell lines after knockdown (Figure 6A-B), which is similar to the findings observed after ARID5B knockdown.13 A similar effect was observed in 2 other T-ALL cell lines, CTV-1 and HSB-2, that express TAL1 and ARIEL (supplemental Figure 6A-B). Importantly, no effects were found in the TAL1+ T-ALL cells that do not express ARIEL (CCRF-CEM and RPMI 8402) after ARIEL shRNA transduction (supplemental Figure 6C-D), supporting the observation that the growth inhibition was specific to the depletion of ARIEL. We also observed a significant decrease in the colony formation ability of Jurkat cells with ARIEL knockdown compared with that in the control cells (supplemental Figure 6E-F).
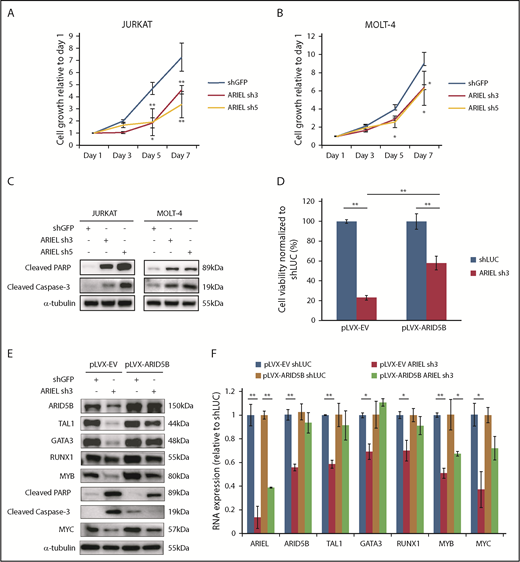
ARIEL is required for T-ALL cell survival in culture. (A-B) Jurkat (A) and MOLT-4 (B) cells were transduced by lentivirus infection with either a control shRNA (shGFP) or an shRNA targeting ARIEL (sh3 and sh5). Cell viability was measured for up to 7 days. Cell growth rates relative to the readings on day 1 are shown as the mean plus or minus SD of triplicate experiments. (C) Western blot analysis was performed using antibodies against cleaved PARP and cleaved caspase 3 in Jurkat and MOLT-4 cells after the shRNA knockdown of ARIEL. (D) Jurkat cells overexpressing empty vector (pLVX-EV) or ARID5B (pLVX-ARID5B) were transduced by lentivirus infection with either a control shRNA (shLUC) or shRNA targeting ARIEL (sh3). Cell viability was measured 5 days after transduction and percentage of viable cells relative to shLUC are shown as the mean plus or minus SD of duplicate experiments. (E) Western blot analysis showing the protein expression of each member of the TAL1 complex and MYC after knockdown of ARIEL in pLVX-EV and pLVX-AIRD5B–overexpressing Jurkat cells. (F) The RNA expressions of ARID5B and each member of the TAL1 complex and MYC were measured by qRT-PCR after the shRNA knockdown of ARIEL in pLVX-EV and pLVX-AIRD5B overexpressing Jurkat cells.
ARIEL is required for T-ALL cell survival in culture. (A-B) Jurkat (A) and MOLT-4 (B) cells were transduced by lentivirus infection with either a control shRNA (shGFP) or an shRNA targeting ARIEL (sh3 and sh5). Cell viability was measured for up to 7 days. Cell growth rates relative to the readings on day 1 are shown as the mean plus or minus SD of triplicate experiments. (C) Western blot analysis was performed using antibodies against cleaved PARP and cleaved caspase 3 in Jurkat and MOLT-4 cells after the shRNA knockdown of ARIEL. (D) Jurkat cells overexpressing empty vector (pLVX-EV) or ARID5B (pLVX-ARID5B) were transduced by lentivirus infection with either a control shRNA (shLUC) or shRNA targeting ARIEL (sh3). Cell viability was measured 5 days after transduction and percentage of viable cells relative to shLUC are shown as the mean plus or minus SD of duplicate experiments. (E) Western blot analysis showing the protein expression of each member of the TAL1 complex and MYC after knockdown of ARIEL in pLVX-EV and pLVX-AIRD5B–overexpressing Jurkat cells. (F) The RNA expressions of ARID5B and each member of the TAL1 complex and MYC were measured by qRT-PCR after the shRNA knockdown of ARIEL in pLVX-EV and pLVX-AIRD5B overexpressing Jurkat cells.
We then analyzed the apoptosis phenotype. We found that knockdown of ARIEL in Jurkat and MOLT-4 cells was accompanied by increased levels of cleaved poly(ADP-ribose) polymerase (PARP) and cleaved caspase-3, which are apoptosis markers (Figure 6C). The same results were also observed in the CTV-1 and HSB-2 cells (supplemental Figure 6G). Additionally, we detected a significant increase in the percentage of cells at the sub-G0/G1 phase of the cell cycle among the cells with the ARIEL knockdown (supplemental Figure 6H-I). Thus, the growth inhibitory effect following ARIEL knockdown was likely due to apoptotic cell death, which is consistent with the phenotype observed after ARID5B knockdown.13 To further confirm this phenotype, we knocked down ARIEL in Jurkat cells that overexpresses the antiapoptotic protein BCL2. In this setting, we observed that overexpression of BCL2 was able to significantly reduce cell death that was induced upon ARIEL knockdown, when compared with the control cells, which was accompanied by decrease in cleaved PARP and caspase 3 (supplemental Figure 6J-K). Notably, the reduction in gene expression of ARID5B and members of the TAL1 complex did not recover in the BCL2-overexpressing cells (supplemental Figure 6K). This suggested that ARIEL’s effect on expression of these genes is specific and not due to excessive cell death.
Moreover, we performed a rescue experiment in Jurkat cells that overexpresses ARID5B and found that cell death induced by knockdown of ARIEL was partially rescued in the presence of ectopic expression of ARID5B when compared with control cells (Figure 6D). Importantly, overexpression of ARID5B was able to partially prevent the decrease in expression of the TAL1 complex members at the mRNA and protein levels (Figure 6E-F). In our previous study, we have also demonstrated that ARID5B regulates the MYC oncogene expression.13 Indeed, MYC expression was downregulated upon ARIEL knockdown (Figure 6E-F) and was able to be partially rescued by ARID5B overexpression. Overall, these observations would suggest that the cellular phenotype observed from loss of ARIEL in the cells are mainly due to its direct effect on ARID5B gene expression.
ARIEL is required for leukemia progression in a xenograft model
Finally, we analyzed the effect of ARIEL in a xenograft model. We established a cell line that constitutively expresses the luciferase gene under a ubiquitous promoter (MOLT-4-Luc). Then, the cells were transduced with shRNA targeting GFP (control), ARIEL, or ARID5B by lentivirus infection and injected into the NSG immunocompromised mice. Engraftment and disease progression were analyzed by luminescence imaging. In this analysis, we observed an expansion of leukemia cells in the control group of mice injected with MOLT-4-Luc cells transduced with shGFP (n = 4) (Figure 7). Importantly, the group of mice injected with MOLT-4-Luc cells transduced with shRNA against ARIEL or ARID5B demonstrated very minimal disease progression (n = 4).
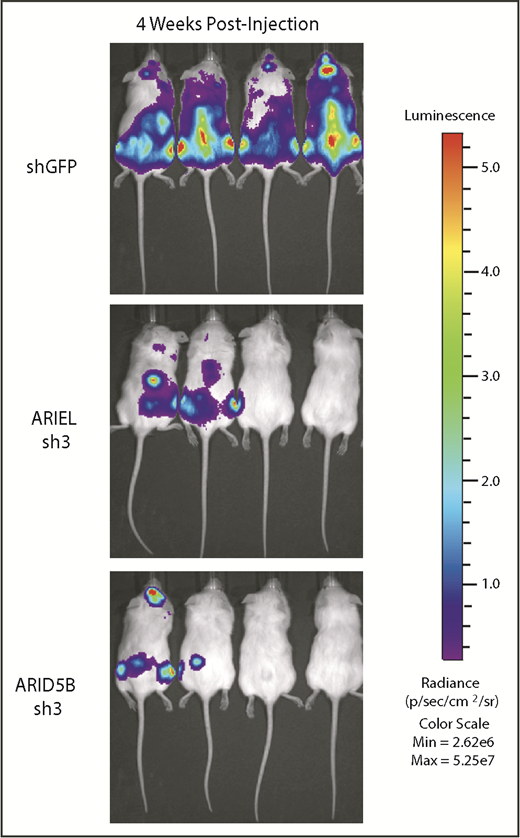
ARIEL is required for T-ALL leukemia progression in a xenograft model. MOLT-4 cells stably expressing the luciferase gene were transduced by lentivirus infection with an shRNA targeting control (shGFP), ARIEL (sh3), or ARID5B (sh3). The cells were injected into NSG mice. Lumino imaging was conducted 4 weeks after the injection.
ARIEL is required for T-ALL leukemia progression in a xenograft model. MOLT-4 cells stably expressing the luciferase gene were transduced by lentivirus infection with an shRNA targeting control (shGFP), ARIEL (sh3), or ARID5B (sh3). The cells were injected into NSG mice. Lumino imaging was conducted 4 weeks after the injection.
Discussion
In our previous study, we profiled lncRNAs expressed in T-ALL cells.34 From this analysis, we identified XLOC_005968/ARIEL, which has not been annotated in normal or cancer cells. The selection of ARIEL for the subsequent analysis was also justified because ARIEL is highly activated under the ARID5B superenhancer specifically in TAL1+ T-ALL cells. We recently reported that ARID5B positively regulates the expression of members of the TAL1 complex, their targets, and the MYC oncogene, thus reinforcing the oncogenic regulatory program in T-ALL.13 Thus, we hypothesized that ARIEL may also play a role in the regulatory network. However, determining whether this lncRNA had particular biological functions was important because it could be simply a by-product of active transcription from the enhancer region. Hence, we performed several functional analyses in this study. Critically, we demonstrated that ARIEL plays a crucial role in bridging the ARID5B enhancer and promoter regions. We have shown that ARIEL can physically interact with the mediator 12 protein and is localized to the ARID5B promoter region. The loss of ARIEL disrupted the enhancer-promoter interaction and reduced the expression of ARID5B in the T-ALL cells. Our results clearly show that ARIEL functions as an eRNA. Of note, our gene expression analysis also suggested that ARIEL might have effects independent of affecting ARID5B and the TAL1 complex. As a future plan, a ChIRP-seq assay would be ideal to comprehensively uncover binding regions of ARIEL to genomic elements.
The role of ARIEL as an eRNA was further supported by the fact that the depletion of ARIEL did not significantly affect the H3K4me1 and H3K27ac active histone markers at the enhancer regions. Thus, the recruitment of histone methyltransferase or acetylase to the ARID5B enhancer most likely does not involve ARIEL. We propose the following model: (1) the TAL1 complex binds the ARID5B −135-kb enhancer and initiates the transcription of ARIEL at this cis-regulatory element, (2) ARIEL binds and recruits the mediator complex to the ARID5B enhancer locus to facilitate enhancer-promoter looping, and (3) the transcription of ARID5B is activated. Of note, other groups previously reported that TAL1 is required for chromatin looping in erythroid cells.38 The authors showed by ChIP-PCR experiments that TAL1 binds both at the locus control region and promoter of the γ-globin gene in erythroid cells, which were reduced after TAL1 knockdown and increased after TAL1 overexpression. Subsequent studies indicated that forced expression and directing of Lim domain binding 1 (LDB1), a cofactor of the TAL1 complex, to the enhancer site can establish enhancer-promoter interaction in the absence of mediator protein.39,40 Interestingly, the authors showed that LDB1 protein physically interacts with mediator 12 but not mediator 1 protein,40 which is similar to the action of ARIEL in T-ALL cells. In contrast, in our study, we observed a DNA binding of the TAL1 complex at the ARID5B −135-kb enhancer, which was reduced after ARIEL knockdown; however, DNA binding of TAL1 was not detected at or near the ARID5B promoter element. Although it is still possible that LDB1 mediates the chromatin looping, our current data suggest that ARIEL is the primary mechanism that specifically mediates the enhancer-promoter interaction.
Furthermore, intragenic enhancers have been shown to act as alternative promoters within protein-coding genes and produce RNA transcripts that are multiexonic and polyadenylated.41,42 Recently, the act of transcription alone at intragenic enhancers has been suggested to attenuate host gene expression by interrupting productive elongation independent of the eRNA transcript being produced.43 However, in our study, the host gene CABCOCO1/C10ORF107 is not expressed in T-ALL, but the intragenic ARID5B −135-kb enhancer actively transcribed ARIEL. To the best of our knowledge, our report is the first to show that this type of ncRNA, which is expressed from intragenic enhancers, has biological functions in T-ALL and is not merely a transcription by-product. Therefore, our study provides novel insight into the molecular functions of lncRNAs.
Of note, we have tried to knock down ARIEL by shRNA transduction in primary T-ALL cells expanded by patient-derived mouse xenograft, to confirm the findings in cell lines. We tested several different protocols, including the IL-7 surface-engineered lentiviral vector and fibronectin-coated plates with a concentrated virus media reported by others.44-47 However, despite numerous attempts, we were not able to efficiently transduce shRNA into the T-ALL patient-derived mouse xenograft cells, although several groups reported successful infection of lentivirus into primary T-ALL samples.44-47 This could be due to the nature of primary human T cells or difference in cell population or methodology. Because this is a major limitation in T-ALL research, we would like to investigate this point as a future plan.
Importantly, our study also demonstrated, for the first time, the involvement of a lncRNA in the regulation of the TAL1-induced oncogenic regulatory program in T-ALL. We have shown that ARIEL is directly regulated by the TAL1 complex, and its expression is higher in TAL1+ cases than in other T-ALL subgroups. Notably, ARIEL is not activated in normal immature thymocytes. Thus, the expression of ARIEL in T-ALL cells is aberrant. In this regard, we have analyzed genetic abnormalities at the ARIEL and ARID5B gene loci using T-ALL cell lines and primary samples used in this study. However, there was no structural variations or genetic mutations that potentially contribute to the abnormal expression of ARIEL (data not shown). Hence, we think that ectopic expression of TAL1 due to its genetic abnormalities followed by its binding at the ARID5B enhancer is the primary determinant that leads to the expression of ARIEL-ARID5B in TAL1+ T-ALL cells. Critically, ARIEL can indirectly regulate the expression of TAL1, its regulatory partners, and its downstream targets by activating ARID5B, thereby contributing to T-cell leukemogenesis. ARIEL knockdown inhibited leukemia cell survival and progression, which was also observed after ARID5B knockdown. Our current study further supports our previous findings implicating ARID5B as a pro-oncogenic factor in T-ALL. The positive feedback loop between ARID5B and ARIEL could be an important “hub” underlying the molecular pathogenesis of TAL1+ T-ALL.
The RNA-seq after ARIEL knockdown data reported in this article have been deposited in the Gene Expression Omnibus (GEO) database (accession number GSE112077).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Nature Publishing Group Language Editing for editing the manuscript. The authors thank members of the Sanda laboratory for discussions and Shi Hui Tan for giving advice on statistical analysis.
This work was supported by the National Research Foundation (NRF) Singapore and the Singapore Ministry of Education (MOE) under its Research Centres of Excellence initiative. The study was also supported by the NRF under its Competitive Research Programme (NRF-NRFF2013-02 [T.S.] and NRF-NRFF2012-054 [M.J.F.]) and the RNA Biology Center at Cancer Science Institute (CSI) Singapore, National University of Singapore (NUS), as part of the funding under the Singapore MOE’s Research Fund Tier 3 (MOE2014-T3-1-006; D.G.T.), as well as a Singapore Translational Research (STaR) Investigator Award (D.G.T.).
Authorship
Contribution: S.H.T., W.Z.L., and F.C.B. performed the experiments; P.C.T.N. and T.K.T. analyzed the RNA-seq and ChIP-seq data sets; O.A., M.C.L., and M.J.F. supervised the 4C assays; Z.L. and A.E.J.Y. provided the primary samples; S.H.T., P.C.T.N., D.G.T., and T.S. designed the study; and S.H.T., D.G.T., and T.S. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Takaomi Sanda, Cancer Science Institute of Singapore, National University of Singapore, 14 Medical Dr, Centre for Translational Medicine, #12-01, Singapore, 117599; e-mail: takaomi_sanda@nus.edu.sg.
