

Key Points
GSK3 is overexpressed in, and functionally exploited by, lymphoma cells.
New GSK3 inhibitor 9-ING-41 induces apoptosis and cell cycle arrest at prophase by targeting centrosomes and microtubule-bound GSK3β.

Abstract
Targeting the B-cell receptor and phosphatidylinositol 3-kinase/mTOR signaling pathways has shown meaningful, but incomplete, antitumor activity in lymphoma. Glycogen synthase kinase 3 (GSK3) α and β are 2 homologous and functionally overlapping serine/threonine kinases that phosphorylate multiple protein substrates in several key signaling pathways. To date, no agent targeting GSK3 has been approved for lymphoma therapy. We show that lymphoma cells abundantly express GSK3α and GSK3β compared with normal B and T lymphocytes at the messenger RNA and protein levels. Utilizing a new GSK3 inhibitor 9-ING-41 and by genetic deletion of GSK3α and GSK3β genes using CRISPR/CAS9 knockout, GSK3 was demonstrated to be functionally important to lymphoma cell growth and proliferation. GSK3β binds to centrosomes and microtubules, and lymphoma cells treated with 9-ING-41 become arrested in mitotic prophase, supporting the notion that GSK3β is necessary for the progression of mitosis. By analyzing recently published RNA sequencing data on 234 diffuse large B-cell lymphoma patients, we found that higher expression of GSK3α or GSK3β correlates well with shorter overall survival. These data provide rationale for testing GSK3 inhibitors in lymphoma patient trials.
Introduction
Lymphoma is the sixth most common cancer,1 and diffuse large B-cell lymphoma (DLBCL) remains the most common non-Hodgkin lymphoma (NHL). Although therapies have improved, there remain unmet needs for the DLBCL and T-cell non-Hodgkin lymphoma (TCL) patients not currently cured and most mantle cell lymphoma (MCL) patients who still die from their disease. Recent research has focused on targeting hyperactivated signal pathways that are essential to lymphoma cell growth.2 Initial success with this strategy has been demonstrated with the Bruton tyrosine kinase inhibitors ibrutinib3 and acalabrutinib,4 the phosphatidylinositol 3-kinase (PI3K) inhibitors idelalisib5 and copanlisib,6 the mTORC1 inhibitor everolimus,7 and the immune-modulatory agent lenalidomide.8 However, the single-agent/single-target strategy often leads to the development of drug resistance. Thus, multiagent regimens have been developed with increased response rates but at higher cost and with more side effects. There remains a need for new agents that target multiple signaling pathways simultaneously.
Glycogen synthase kinase 3 (GSK3) α and β are highly homologous and functionally overlapping serine/threonine kinases encoded by independent genes GSK3A and GSK3B, respectively. Given their functional redundancy, we refer to both as GSK3 unless otherwise specified. GSK3 phosphorylates >100 protein substrates that are components of many key cellular pathways, including Wnt/β-catenin, Hedgehog, Notch, NF-κB, and PI3K/Akt9-11 ; therefore, GSK3 has a widespread potential impact on health and disease. Furthermore, PI3K/AKT/GSK3β signaling also governs glucose hypermetabolism in cancer cells.12 A characteristic feature of all aggressive lymphoma types and relapsed low-grade NHL is increased glucose metabolism, as vividly demonstrated by 2-deoxy-2-[18F] fluoro-d-glucose–positron emission tomography imaging. Targeting pathways of glucose hypermetabolism offers potential for therapeutic benefit in NHL.13
We hypothesized that targeting GSK3 may be a new strategy in lymphoma treatment by interfering with tumor growth and survival through pathways not affected by standard therapies. The purpose of this study is to understand whether GSK3α and GSK3β are upregulated in lymphoma cells, determine whether these kinases are essential to lymphoma cell survival, and whether inhibiting GSK3 results in any antilymphoma activity. We also investigate the potential mechanisms by which GSK3 inhibitors kill lymphoma cells and explore the therapeutic potential for a novel GSK3 inhibitor 9-ING-41.
Materials and methods
Primary lymphoma cells and lymphoma cell lines
Lymphoma tissue samples were obtained from patients after written informed consent. This Lymphoma Specialized Program of Research Excellence (SPORE) biospecimens protocol was approved by the Mayo Clinic Institutional Review Board in accordance with the Declaration of Helsinki. All primary patient samples were biopsy tissues from spleen or lymph nodes. Fresh tissue samples were gently dissociated into cell suspension and subjected to Ficoll-Paque density gradient centrifugation. Primary lymphoma cells were then used directly for proliferation assay or stored at −80°C for western blot analysis after lymphoma diagnosis confirmation.
All lymphoma cell lines used in this study were purchased from the American Type Culture Collection (Manassas, VA) or German Collection of Microorganisms and Cell Cultures GmbH (Braunschweig, Germany). DLBCL lines were cultured in Iscove modified Dulbecco medium supplemented with 10% human serum (Sigma-Aldrich); TCL and MCL lines were maintained in RPMI 1640 medium supplemented with 10% fetal calf serum. The Jeko cell line used for xenograft modeling in mice was stably expressed with firefly luciferase (Fluc) through lentiviral transduction. Cell lines are periodically checked for the absence of mycoplasma infection and authenticated by a home brew single nucleotide polymorphism–based polymerase chain reaction (PCR) method or short tandem repeat profiling through the American Type Culture Collection.
CRISPR/CAS9 approach to deleting GSK3α and/or GSK3β
Guide RNAs (gRNAs) for targeting the first coding exons of both GSK3α and GSK3β genes were designed using a Web tool (http://crispr.mit.edu/). The gRNA sequences (GSK3α: GACAGATGCCTTTCCGCCGC; GSK3β: CGGCTTGCAGCTCTCCGCAA) were cloned into the px458 vector (Addgene) carrying a coexpressing GFP. The constructs were nucleofected into lymphoma cells using a nucleofection kit (Lonza, Basel, Switzerland). Thirty-six hours postnucleofection, GFP-expressing single cells were sorted into 96-well plates at 1 cell per well on an FACSAria II sorter. After the expansion of single-cell subclones in culture for 2 weeks, each subclone was genotyped by PCR and Sanger’s DNA sequencing.
Lymphoma xenograft modeling in mice
All mouse work was conducted in compliance with the Mayo Clinic Institutional Animal Care and Use Committee protocol. NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ) mice used in our experiments were purchased from the Jackson Laboratory (Bar Harbor, ME). Eight to ten mice of the same sex were subcutaneously injected at 8 weeks of age with 5 × 106 Fluc-expressing Jeko cells in the right flanks. Tumor engraftment was verified by imaging 4 days after Jeko cell inoculation. The tumor-engrafted mice were randomly grouped into control and treatment groups and were left untreated or treated with 9-ING-41 by intraperitoneal injection, as indicated. Tumor volumes were measured with an IVIS Imager (Xenogen, Alameda, CA) 20 minutes after intraperitoneal injection of 200 μL of 15 mg/mL D-Luciferin (GoldBio, St. Louis, MO) and anesthesia with 2.5% isoflurane. All imaging variables were kept consistent for comparativeness. The experiment was terminated when the largest tumor met the size limit of the Institutional Animal Care and Use Committee protocol.
Detailed information about antibodies and chemicals, as well as methods used for cell apoptosis, proliferation, western blotting, immunofluorescence staining, immunohistochemistry staining, reverse-transcription quantitative PCR, cell cycle analysis, and RNA sequencing (RNA-Seq) data analysis are described in supplemental Materials and methods (available on the Blood Web site).
Results
GSK3α and GSK3β are overexpressed in lymphoma cells
To determine whether GSK3 would constitute an effective therapeutic target in lymphoma, we examined the expression status of GSK3α and GSK3β in purified human normal B and T cells and in DLBCL, MCL, and TCL cell lines. By reverse-transcription quantitative PCR, we found that most lymphoma lines showed higher, but variable, levels of GSK3α and GSK3β mRNAs compared with normal lymphocytes (Figure 1A). These results suggest that the transcription of GSK3 is enhanced in most lymphoma cell lines. We next analyzed GSK3 protein expression by western blotting; likewise, most lymphoma lines (with the exception of Ly-19) showed strong expression of GSK3α protein compared with weak expression in B and T lymphocytes (Figure 1B, green). Similarly, GSK3β protein was also strongly expressed in all lymphoma cell lines but very weakly expressed (visible after long exposure; data not shown) in normal lymphocytes (Figure 1B, red). Our data suggest that GSK3 proteins are indeed overexpressed in most B- and T-lymphoma cell lines. However, the levels of mRNA and proteins of a given cell line are not always in perfect agreement. This is not surprising, because the steady-state levels of protein and mRNA are determined by many factors that govern their production, as well as their stability.

GSK3α and GSK3β mRNA and proteins are overexpressed in lymphomas. (A) Real-time PCR quantitation showing that GSK3α and GSK3β mRNAs are overexpressed in lymphoma lines in comparison with low expression in normal B or T lymphocytes. (B) Western blot images demonstrating that GSK3α and GSK3β proteins are also abundantly expressed in various lymphoma lines in comparison with purified normal B or T lymphocytes.
GSK3α and GSK3β mRNA and proteins are overexpressed in lymphomas. (A) Real-time PCR quantitation showing that GSK3α and GSK3β mRNAs are overexpressed in lymphoma lines in comparison with low expression in normal B or T lymphocytes. (B) Western blot images demonstrating that GSK3α and GSK3β proteins are also abundantly expressed in various lymphoma lines in comparison with purified normal B or T lymphocytes.
Because phosphorylation of GSK3α at serine 21 and GSK3β at serine 9 each renders the protein functionally inactive, we wondered whether GSK3α and GSK3β are phosphorylated at these sites in lymphoma cells. By western blotting, both proteins were variably phosphorylated across our lymphoma line panel similar to normal lymphocytes. We did not see any unique pattern that is specific to lymphoma lines (supplemental Figure 1), suggesting that the baseline phosphorylation of GSK3 is unlikely to be significant in lymphoma. Taken together, our results suggest that GSK3α and GSK3β are abundantly expressed in most lymphoma cell lines compared with normal lymphocytes, supporting the potential of targeting GSK3 for therapeutic benefit.
GSK3α and GSK3β are functionally important in lymphoma cells
Given that GSK3α and GSK3β are overexpressed in lymphoma cells and that both enzymes are implicated in multiple signaling pathways critical for cell functions, we examined whether GSK3α and GSK3β are functionally supporting the survival and proliferation of lymphoma cells. To that end, we took 2 complementary approaches: the first used a new GSK3 inhibitor, 9-ING-41, and the second used CRISPR/CAS9 to knock out GSK3A and GSK3B genes. Treatment of TCL and MCL lines with low doses of 9-ING-41 for 48 hours induced apoptosis (Figure 2A); the DLBCL lines required higher concentrations (Figure 2B). In contrast, we did not detect significant apoptosis in purified normal unstimulated T lymphocytes or peripheral blood mononuclear cells, even at a concentration of 10.0 µM of 9-ING-41. We further calculated the inhibitory concentrations of 9-ING-41 at half of the maximal effect (IC50) on cell survival of various lymphoma cell lines (Table 1). Our data indicate that 9-ING-41 can specifically induce lymphoma cell apoptosis without affecting normal lymphocytes.

GSK3 is essential for lymphoma cell proliferation and survival. Unstimulated peripheral blood B and T lymphocytes isolated from a healthy donor were used as normal control. Proapoptotic effect of the GSK3 inhibitor 9-ING-41 in various MCL and TCL lines (A) and in DLBCL lines (B). (C) Cell-proliferation profile of various lymphoma cell lines upon treatment with 9-ING-41. All results are from 3 independent experiments.
GSK3 is essential for lymphoma cell proliferation and survival. Unstimulated peripheral blood B and T lymphocytes isolated from a healthy donor were used as normal control. Proapoptotic effect of the GSK3 inhibitor 9-ING-41 in various MCL and TCL lines (A) and in DLBCL lines (B). (C) Cell-proliferation profile of various lymphoma cell lines upon treatment with 9-ING-41. All results are from 3 independent experiments.
IC50 of 9-ING-41 for cell survival and cell proliferation in various lymphoma cell lines
| Lymphoma line | Cell survival | Cell proliferation |
|---|---|---|
| DLBCL | ||
| OCI-LY1 | 3.05 | 0.69 |
| OCI-LY19 | 2.21 | 1.96 |
| OCI-Ly3 | 3.76 | 1.03 |
| SU-DHL6 | 1.58 | 0.84 |
| MCL | ||
| Granta-519 | 0.77 | 0.38 |
| Jeko | 1.28 | 0.94 |
| Mino | 0.55 | 0.72 |
| TCL | ||
| Karpas-299 | 3.34 | 0.26 |
| MyLa | 0.69 | 0.19 |
| SeAx | 1.60 | 0.38 |
| Lymphoma line | Cell survival | Cell proliferation |
|---|---|---|
| DLBCL | ||
| OCI-LY1 | 3.05 | 0.69 |
| OCI-LY19 | 2.21 | 1.96 |
| OCI-Ly3 | 3.76 | 1.03 |
| SU-DHL6 | 1.58 | 0.84 |
| MCL | ||
| Granta-519 | 0.77 | 0.38 |
| Jeko | 1.28 | 0.94 |
| Mino | 0.55 | 0.72 |
| TCL | ||
| Karpas-299 | 3.34 | 0.26 |
| MyLa | 0.69 | 0.19 |
| SeAx | 1.60 | 0.38 |
All data are μM.
To test the role of GSK3 in lymphoma cell proliferation, we performed a thymidine-incorporation assay in the presence or absence of 9-ING-41. The proliferation rate of all TCL and MCL lines was profoundly inhibited in the presence of 9-ING-41 concentrations as low as 1.0 µM; the DLBCL lines required slightly higher concentrations. We also calculated the IC50 of 9-ING-41 on cell proliferation for various lymphoma cell lines (Table 1). Our data suggest that GSK3 activity is indeed important for the proliferation and survival of lymphoma cells.
In a second approach, we used a CRISPR/CAS9 knockout technique to genetically delete GSK3A and GSK3B genes. After transient expression of the construct carrying CAS9-T2A-GFP and gRNA-specific GSK3A or GSK3B gene exon 1 sequences, GFP-expressing single cells were sorted into a 96-well plate by flow sorting. After 2 to 3 weeks in culture, single-cell subclones carrying a unique modification in GSK3A, GSK3B, or both genes were genotyped for the gene deletion and verified by western blot for the protein depletion. As summarized in Table 2, several GSK3A-null knockout subclones were readily obtained from all 5 cell lines tested, and GSK3B-null subclones were also obtained from Ly-1 cell lines. However, after analyzing 24 to 36 single-cell subclones from Ly-19, Jeko, Mino, and Karpas 299 cell lines, no knockout subclones were detected (ie, all surviving clones were wild-type or carried heterozygous mutations, suggesting that GSK3B-null cells from those cell lines likely died during culture). Given that CRISPR/CAS9 is a highly efficient approach for biallelic deletion of the GSK3B gene in Ly-1 cells (19/24; 79%) but not GSK3B-null clones in any other lymphoma lines tested (0/24, 0/24, 0/36, 0/26; 0%), our data support the notion that GSK3B is necessary for the survival of lymphoma cells in these cell lines. Using short hairpin RNA knockdown, we observed similar results, showing that GSK3B knockdown is lethal in several lymphoma lines with the exception of Ly-1 (data not shown). Interestingly, unlike other lines that all died following GSK3B knockout, we derived multiple Ly-1 subclones with homozygous deletion of GSK3A, GSK3B, or both genes. Thus, Ly-1, a germinal center B-cell–like type DLBCL cell line, likely possesses a unique unexplained compensatory mechanism(s) that prevents the cells from dying upon GSK3B deletion. Taken together, we have shown pharmacologically and genetically that GSK3B function is indeed necessary for the proliferation and survival of lymphoma cells. This suggests that targeting GSK3B has potential in lymphoma treatment.
Effect of GSK3α and/or GSK3β knockout using CRISPR/Cas9 approach on the survival of various lymphoma cell lines
| Lymphoma line | Null-knockout clones/total clones screened | ||
|---|---|---|---|
| GSK3α | GSK3β | GSK3α/β | |
| OCI-LY1 | 9/17 | 19/24 | 6/13 |
| OCI-LY19 | 20/28 | 0/24 | nd |
| Jeko | 7/12 | 0/24 | nd |
| Mino | 5/12 | 0/36 | nd |
| Karpas299 | 12/20 | 0/26 | nd |
| Lymphoma line | Null-knockout clones/total clones screened | ||
|---|---|---|---|
| GSK3α | GSK3β | GSK3α/β | |
| OCI-LY1 | 9/17 | 19/24 | 6/13 |
| OCI-LY19 | 20/28 | 0/24 | nd |
| Jeko | 7/12 | 0/24 | nd |
| Mino | 5/12 | 0/36 | nd |
| Karpas299 | 12/20 | 0/26 | nd |
nd, not done.
GSK3 inhibition blocks G2/M progression in lymphoma cells
Our finding that 9-ING-41 strongly inhibits proliferation in vitro prompted us to examine the effect of 9-ING-41 on lymphoma cell cycle kinetics. After 9-ING-41 treatment, we consistently observed cell cycle blockage at G2/M after as little as 24 hours of treatment in all lines tested (Figure 3A), suggesting that GSK3 activity is required for successful progression of mitosis. To further determine whether this G2/M arrest specifically resulted from GSK3β inhibition, we examined the cell cycle profile of parental (wild-type) and GSK3A-, GSK3B-, and GSK3A/B-knockout Ly-1 subclones. In the absence of treatment, parental wild-type and GSK3A-null Ly-1 cells showed normal cell cycle profiles, whereas GSK3B- and GSK3A/B-knockout subclones exhibited increased cells in G2/M (Figure 3B). In addition, GSK3A/B double-knockout subclones showed increased levels of polyploid (>4N) cells, possibly as a result of defective mitosis. These cell cycle abnormalities of GSK3B-null and GSK3A/B-null Ly-1 cells are subtle and have little impact on the survival of the Ly-1 progeny, likely as a result of Ly-1 cell line–specific compensatory mechanisms. In summary, 9-ING-41 treatment phenocopies the effect of GSK3B single or GSK3A/B double deletion on cell cycle progression, suggesting that GSK3B is critical for lymphoma cell cycle G2/M progression, and 9-ING-41 is a potent cell cycle–blocking agent for lymphoma cells.
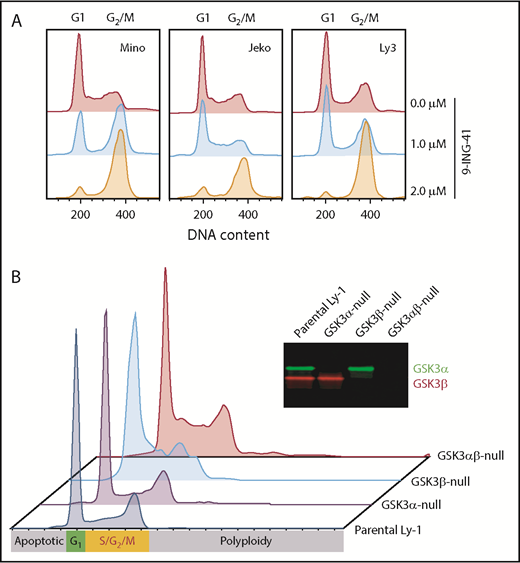
GSK3 inhibition or deletion in lymphoma cells leads to cell cycle arrest in G2/M. (A) Cell cycle profile of 3 representative cell lines Jeko, Mino, and OCI-Ly3 after a 24-hour treatment with 0, 1.0, or 2.0 μM 9-ING-41. (B) Cell cycle profiles of parental Ly-1 cells and GSK3α-, GSK3β-, and GSK3αβ-knockout subclones. Inset: western blot image showing the depletion of GSK3α and GSK3β protein in the knockout Ly-1 subclones.
GSK3 inhibition or deletion in lymphoma cells leads to cell cycle arrest in G2/M. (A) Cell cycle profile of 3 representative cell lines Jeko, Mino, and OCI-Ly3 after a 24-hour treatment with 0, 1.0, or 2.0 μM 9-ING-41. (B) Cell cycle profiles of parental Ly-1 cells and GSK3α-, GSK3β-, and GSK3αβ-knockout subclones. Inset: western blot image showing the depletion of GSK3α and GSK3β protein in the knockout Ly-1 subclones.
GSK3 inhibition arrests lymphoma cells at the prophase stage of mitosis
Although cells arrested in G2/M appear as a single DNA content (4N) peak on a flow cytometry graph (Figure 3A), there are actually ≥5 sequential steps (M1-M5) in G2/M that are critical to successful cell division. These include prophase (M1; chromosome condensation, mitotic spindle formation starts), prometaphase (M2; nuclear membrane breakdown, centrosome polarization), metaphase (M3; chromosome pairs align middle plane), anaphase (M4; daughter chromatids separate), telophase (M5; reformation of daughter nuclei), and the final cytokinesis (separation of 2 daughter cells).14 Each of these discrete steps has a unique identifiable nuclear morphology on Wright’s stained cells (Figure 4A). We examined the morphology of untreated and 9-ING-41–treated Jeko cells to determine at what stage they become arrested. As shown in Figure 4B (left panel), all M1 to M5 mitotic steps were readily identified in untreated mitotic Jeko cells; however, in 9-ING-41–treated cells (right panel), a large fraction of cells exhibited condensed chromosomes and reduced cytoplasmic staining resembling prophase (M1) cells without identifiable cells with M2 to M5 morphology. By differential counting of 100 mitotic cells, all stages (M1-M5) of mitotic cells were readily identifiable in untreated cells; only prophase (M1) cells were accounted for in 9-ING-41–treated mitotic cells (Figure 4C). Similar results (data not shown) were observed in other lymphoma lines, including DHL-6, Ly-3, Mino, and Karpas 299. These observations support the conclusion that GSK3 activity is necessary for the progression of mitotic prophase.
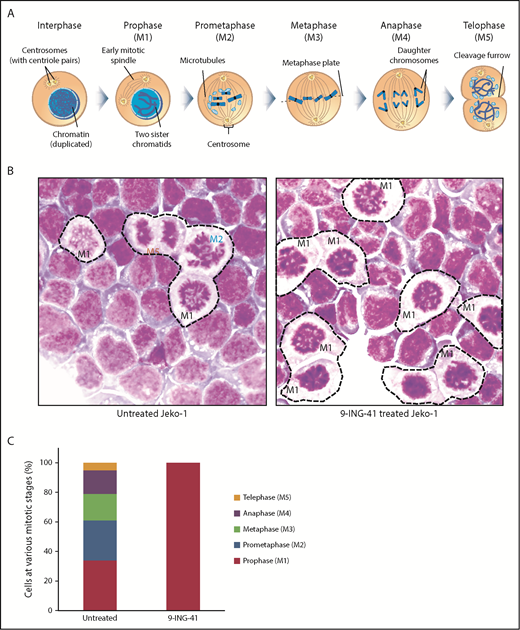
Inhibition of GSK3 by 9-ING-41 leads to mitotic prophase arrest. (A) Cartoon depiction of the sequential steps (M1-M5) during mitosis (purchased from Shutterstock and modified). (B) Representative images of Wright’s stained Jeko cells that were left untreated or treated with 1.0 μM 9-ING-41 for 24 hours. Various mitotic stage cells (M1-M5) are readily identified in untreated cells (left panel), whereas large numbers of only prophase cells (M1) are seen in 9-ING-41–treated cells (right panel); original magnification ×600. (C) Bar chart showing the number of mitotic M1 to M5 cells identified when 100 untreated or 9-ING-41–treated Jeko cells were counted. Similar results (data not shown) were observed in ≥4 lymphoma cell lines.
Inhibition of GSK3 by 9-ING-41 leads to mitotic prophase arrest. (A) Cartoon depiction of the sequential steps (M1-M5) during mitosis (purchased from Shutterstock and modified). (B) Representative images of Wright’s stained Jeko cells that were left untreated or treated with 1.0 μM 9-ING-41 for 24 hours. Various mitotic stage cells (M1-M5) are readily identified in untreated cells (left panel), whereas large numbers of only prophase cells (M1) are seen in 9-ING-41–treated cells (right panel); original magnification ×600. (C) Bar chart showing the number of mitotic M1 to M5 cells identified when 100 untreated or 9-ING-41–treated Jeko cells were counted. Similar results (data not shown) were observed in ≥4 lymphoma cell lines.
GSK3β is localized to centrosomes and mitotic spindles of lymphoma cells
Knowing that GSK3 inhibition leads to mitotic arrest at prophase, we next questioned the involvement of GSK3β in 2 key prophase events: centrosome polarization and mitotic spindle formation. We examined the subcellular localization of GSK3β protein in interphase Jeko or Ly-1 cells by immunofluorescence staining. During interphase, GSK3β is prominently localized in the nucleus and centrosome-like pair dots in the cytoplasm of Jeko cells (Figure 5A-B). To further demonstrate that those cytoplasmic pair dots were indeed centrosomes, we first costained wild-type Ly-1 cells for GSK3β protein and the centrosome marker pericentrin using an incomplete fixation protocol and found that the cytoplasmic GSK3β dots were perfectly colocalized with pericentrin (Figure 5D-F), suggesting that these GSK3β bright dots (Figure 5A-C,F) are indeed centrosomes. We further demonstrated that the anti-GSK3β antibody staining was specific to GSK3β protein by showing its absence in GSK3B-null Ly1 cells (Figure 5G-J). Therefore, we conclude that GSK3β is localized to centrosomes and the nucleus in interphase cells.
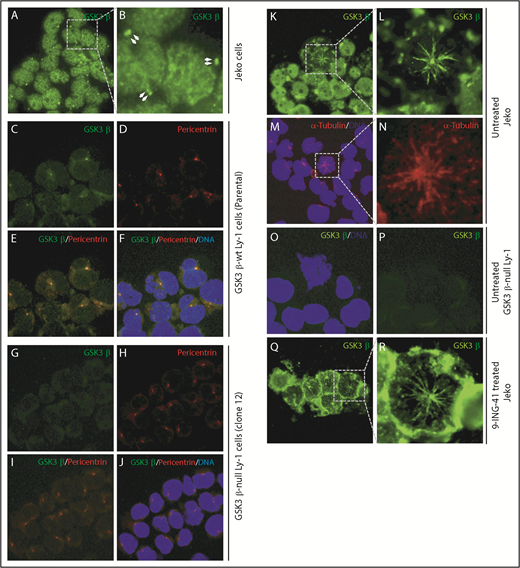
GSK3β localized to centrosomes. (A) Immunofluorescence image showing GSK3β is localized to the nucleus and centrosome pairs in interphase Jeko cells. (B) Enlarged image of the boxed area in panel A. Centrosome pairs are marked by arrows. (C-F) Single- or multichannel images of coimmunostaining of GSK3β and pericentrin in wild-type Ly-1 cells showing their colocalization to the centrosomes. (G-J) Single- or multichannel images of coimmunostaining of GSK3β and pericentrin in GSK3β-null Ly-1 cells showing that staining is specific to GSK3β. (K) Immunofluorescence image showing GSK3β (green) localized to firework-like structures resembling mitotic spindles in mitotic Jeko cells. (L) Enlarged image of the mitotic cell shown in panel K. (M) Overlay image showing the microtubule structure by α-tubulin (red) staining and DNA (blue). (N) Enlarged image of the boxed area in panel M. (O-P) Images showing that spindle structure staining of GSK3β is absent in GSK3β-null Ly-1 cells. (Q) Immunofluorescence image showing GSK3β localized to mitotic spindle structure and polarized centrosomes in 9-ING-41–treated Jeko cells. (R) Enlarged image of the representative mitotic cell shown in panel Q. Original magnification ×600.
GSK3β localized to centrosomes. (A) Immunofluorescence image showing GSK3β is localized to the nucleus and centrosome pairs in interphase Jeko cells. (B) Enlarged image of the boxed area in panel A. Centrosome pairs are marked by arrows. (C-F) Single- or multichannel images of coimmunostaining of GSK3β and pericentrin in wild-type Ly-1 cells showing their colocalization to the centrosomes. (G-J) Single- or multichannel images of coimmunostaining of GSK3β and pericentrin in GSK3β-null Ly-1 cells showing that staining is specific to GSK3β. (K) Immunofluorescence image showing GSK3β (green) localized to firework-like structures resembling mitotic spindles in mitotic Jeko cells. (L) Enlarged image of the mitotic cell shown in panel K. (M) Overlay image showing the microtubule structure by α-tubulin (red) staining and DNA (blue). (N) Enlarged image of the boxed area in panel M. (O-P) Images showing that spindle structure staining of GSK3β is absent in GSK3β-null Ly-1 cells. (Q) Immunofluorescence image showing GSK3β localized to mitotic spindle structure and polarized centrosomes in 9-ING-41–treated Jeko cells. (R) Enlarged image of the representative mitotic cell shown in panel Q. Original magnification ×600.
To determine the intracellular localization of GSK3β in mitotic cells, we analyzed GSK3β localization in normal mitotic Jeko cells. GSK3β staining (green, Figure 5K-L) exhibited a “firework-like” pattern, with polarized centrosomes in the middle and mitotic spindles or microtubules extending outward. The staining is specific to GSK3β, because such staining is absent in GSK3B-deficient mitotic cells (Figure 5O-P). Using a separate stain for the microtubule marker α-tubulin, we show that α-tubulin exhibits a staining pattern similar to that of GSK3β (red, Figure 5M-N), suggesting that GSK3β is localized to microtubules during mitosis. These observations collectively suggest that GSK3β protein is specifically localized to centrosomes and mitotic spindles during mitosis. Next, we determined the localization of GSK3β in 9-ING-41–treated Jeko cells (Figure 5Q-R). We found similar firework-like GSK3β staining patterns in all prophase cells without any altered GSK3β localization. Taken together, these data indicate that GSK3β localized to centrosomes and nucleus during interphase (Figure 5A-B) and to centrosomes and mitotic microtubules during mitosis (Figure 5K-L). Treatment with 9-ING-41 did not alter the localization of GSK3β or affect centrosome polarization or microtubule formation; therefore, 9-ING-41 is likely to inhibit microtubule function at a later step, preventing the progression of prophase.
GSK3 expression and targeting in primary cells from lymphoma patients
Having demonstrated that 9-ING-41 potently inhibits lymphoma proliferation and survival in lymphoma cell lines, we wanted to learn whether this remains true in patient samples. To that end, we examined the expression of GSK3α and GSK3β proteins in primary lymphoma cells freshly isolated from patients with MCL, high-grade B-cell lymphoma, follicular lymphoma grade 3B, DLBCL, or angioimmunoblastic TCL. All 5 samples had stronger expression of GSK3α and GSK3β proteins compared with normal blood B-cell controls (Figure 6A). These patient cells were similarly responsive to the antiproliferative effects of 9-ING-41 (Figure 6B). To complement our data on limited fresh patient samples, we further probed paraffin samples, from patients in a different cohort with various lymphoma types, for GSK3β protein expression by immunohistochemistry. As shown in Figure 6C, we found GSK3β overexpression in all samples, with varying intensity. Similar to our results, RNA-Seq analysis on primary DLBCL patient samples derived from a public database (Gene Expression Profile Interactive Analysis, http://gepia.cancer-pku.cn) also showed increased expression of GSK3α and GSK3β in lymphoma compared with normal lymphocytes.
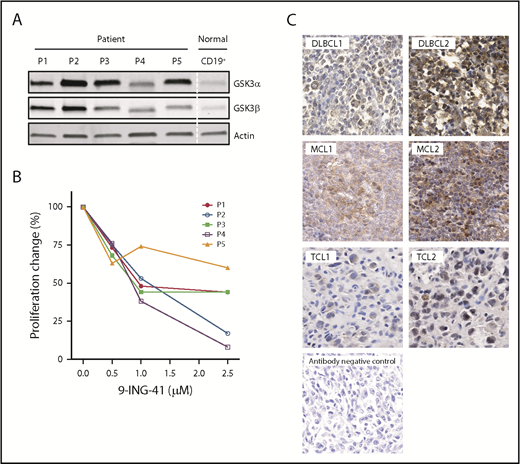
Aberrant expression of GSK3 proteins in primary lymphoma patient (P) cells and their proliferative response to 9-ING-41. (A) Immunoblot of GSK3α and GSK3β proteins showing overexpression in patient samples vs normal B-cell control. P1, MCL; P2, high-grade B-cell lymphoma; P3, follicular large B-cell lymphoma 3B; P4, DLBCL; and P5, angioimmunoblastic T-cell lymphoma. (B) 9-ING-41 inhibited proliferation in all 5 patient samples. (C) Immunohistochemistry staining of GSK3β on paraffin tissue sections from patients with various lymphomas (original magnification ×40). Representative images show the spectrum of GSK3β (brown) overexpression in different lymphoma samples. Methylene blue counterstaining (blue) reveals cells negative for GSK3β in the background and in the antibody negative-control panel.
Aberrant expression of GSK3 proteins in primary lymphoma patient (P) cells and their proliferative response to 9-ING-41. (A) Immunoblot of GSK3α and GSK3β proteins showing overexpression in patient samples vs normal B-cell control. P1, MCL; P2, high-grade B-cell lymphoma; P3, follicular large B-cell lymphoma 3B; P4, DLBCL; and P5, angioimmunoblastic T-cell lymphoma. (B) 9-ING-41 inhibited proliferation in all 5 patient samples. (C) Immunohistochemistry staining of GSK3β on paraffin tissue sections from patients with various lymphomas (original magnification ×40). Representative images show the spectrum of GSK3β (brown) overexpression in different lymphoma samples. Methylene blue counterstaining (blue) reveals cells negative for GSK3β in the background and in the antibody negative-control panel.
Given our findings that GSK3α and GSK3β are overexpressed in lymphoma, we hypothesized that overexpression of either gene product may correlate with a patient’s clinical outcome. To that end, we analyzed recently published RNA-Seq data15 for a cohort of 234 DLBCL patients with clinical survival data (median follow-up was 10.5 years; 95% confidence interval [CI], 7.9-not reached). A receiver operating characteristics curve analysis was performed to dichotomize the GSK3α and GSK3β expression to establish the optimal cutoffs (10.5 log2RPKM for GSK3α and 8.6 for GSK3β) for high- and low-expression grouping. As shown in supplemental Figure 2, the GSK3α high-expression group (≥10.5 log2RPKM, n = 172) had an overall survival (OS) of 7.8 years (95% CI, 7.2-8.4), whereas the low-expression group (<10.5 log2RPKM, n = 62) had a significantly higher OS of 8.9 years (95% CI, 8.2-10.1; P = .03). Similarly, the GSK3β high-expression group (≥8.6 log2RPKM, n = 170) had an OS of 7.8 years (95% CI, 7.2-8.2), whereas the GSK3β low-expression group (<8.6 log2RPKM, n = 62) had a significantly higher OS of 9.7 years (95% CI, 8.6-11.5; P = .0005). The results suggest that overexpression of GSK3α or GSK3β correlates with poorer clinical outcome. In addition, the grouping data show that the majority of DLBCL patients are included in the high-expression group for GSK3α or GSK3β, further validating our conclusion that GSK3α and GSK3β are generally overexpressed in lymphoma.
Targeting GSK3 in mouse xenografts of human lymphoma
Given that the GSK3 inhibitor 9-ING-41 effectively inhibits the proliferation and survival of lymphoma cells in vitro, we wanted to know whether it would also have antilymphoma activity in vivo. To that end, we established an MCL xenograft mouse model by subcutaneously injecting NGS mice with Jeko cells expressing Fluc. In 2 independent experiments, 8 and 10 mice with engrafted tumors verified by imaging (typically 4 days after tumor inoculation) randomly served as controls or received intraperitoneal 9-ING-41 (40 mg/kg) every other day (Figure 7A). As shown in Figure 7B, the control (untreated) mice had large tumors with marked luciferase activities by day 17; however, the 9-ING-41–treated mice had smaller tumors with much lower luciferase activity. Our data demonstrated that 9-ING-41 has single-agent antitumor activity in a mouse model of MCL.
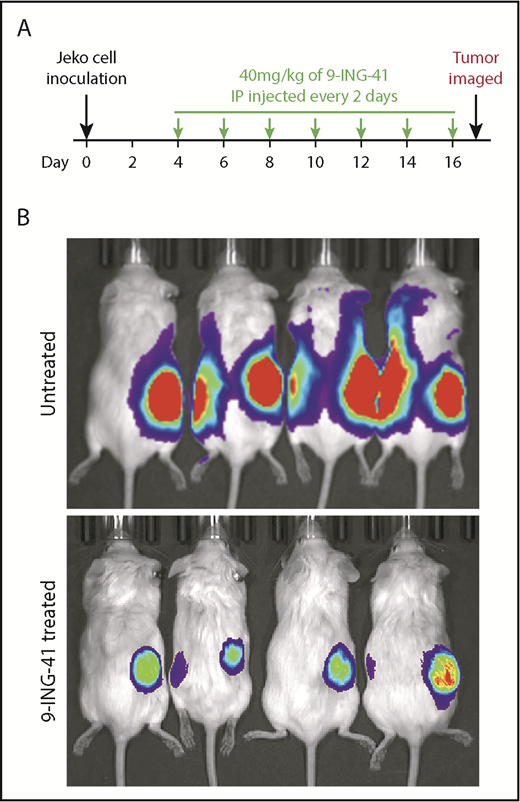
Antilymphoma effect of 9-ING-41 in vivo in Jeko-derived xenograft mouse model. (A) Experimental design showing 9-ING-41 treatment schedule and dosage. (B) Bioluminescence images of xenograft-bearing mice that were left untreated or treated with 9-ING-41. The images were taken at the end of the experiment (day 17). The experiment was performed twice; both showed similar results.
Antilymphoma effect of 9-ING-41 in vivo in Jeko-derived xenograft mouse model. (A) Experimental design showing 9-ING-41 treatment schedule and dosage. (B) Bioluminescence images of xenograft-bearing mice that were left untreated or treated with 9-ING-41. The images were taken at the end of the experiment (day 17). The experiment was performed twice; both showed similar results.
Discussion
GSK3α and GSK3β are functionally related multifunctional serine/threonine protein kinases that are implicated in many signaling pathways that support the proliferation of lymphomas.11 Glucose hypermetabolism is a hallmark of active lymphoma, and GSK3 is a key regulator of glucose metabolism through the PI3K/mTOR and several TRAF3-regulated pathways. Cellular GSK3 status also influences the effects of other kinase inhibitors,16 making GSK3 an attractive target for lymphoma therapeutics.
Herein, we show that GSK3α and GSK3β are overexpressed in lymphoma subtypes considered to be glucose avid. GSK3 activity can be inhibited in vitro with a novel GSK3 inhibitor, 9-ING-41, or by genetic deletion of GSK3α and GSK3β using CRISPR/CAS9. Each approach resulted in effective killing of lymphoma cells. One mechanism of this toxicity that we identified is the requirement for GSK3β in mitotic spindle function. 9-ING-41 treatment induced cell cycle arrest at prophase, likely as a result of defective mitotic spindle function, preventing cell cycle progression to prometaphase. Our data demonstrate that GSK3 is critical for the proliferation and survival of lymphoma in general and MCL and TCL in particular. Karmali et al have also recently reported that 9-ING-41 treatment in vitro can induce apoptosis in a set of lymphoma cell lines different from the ones we studied.17 Their results, in addition to our mechanistic insights into the roles of GSK3 in lymphoma biology, provide rationale for studies in patients with lymphoma.
GSK3α and GSK3β have been reported to be overexpressed in various solid tumors,18-31 and GSK3β overexpression correlates with an inferior prognosis in endometrial cancer.32 Increased intracellular pools of GSK3β have been found in pancreatic cancer cells,33 and nuclear accumulation of GSK3β is associated with poor cell differentiation.28 Consistent with our findings, Pérez-Benavente et al reported that the transcription factor and proto-oncogene JunB is strongly upregulated in anaplastic lymphoma kinase–positive anaplastic large cell TCL, and its phosphorylation is mediated by GSK3β.34 Therefore, it is conceivable that these enzymes may play different roles in different cancers.
There have been other reports of the association of GSK3 with centrosomes. Mbom et al reported that GSK3β and NEK2 phosphorylate β-catenin, which is also localized to mitotic centrosomes.35 GSK3 reduces the stability of securin, an important regulator of mitosis, by phosphorylation and consequent ubiquitination of the protein.36 Lee et al showed that GSK3β physically binds to and phosphorylates hBora, another mitotic factor that is critical for Aurora A kinase to activate entry into mitosis.37 We describe here that GSK3β binds to centrosomes and mitotic spindles, 2 key features of mitotic prophase, and 9-ING-41 treatment leads to cell cycle arrest at mitotic prophase. The exact molecular mechanism of this cell cycle arrest requires further exploration. Furthermore, the effects of 9-ING-41 likely extend well beyond the cell cycle arrest. Indeed, it is well known that other signaling pathways involving GSK3, including Notch,38 Wnt,39 and Hedgehog40 pathways, are critical in many lymphoma subtypes, and key components in these pathways are frequently mutated or aberrantly expressed. Our RNA-Seq data on GSK3-deficient Ly-1 cell clones revealed significantly reduced expression of components of many pathways, including glycolysis (data not shown), confirming our initial hypothesis that multifaceted GSK3 is a promising new multifunctional lymphoma target.
In addition to its role in the proliferation and survival of tumor cells, inhibition of GSK3 has recently been implicated in modulating the function of CD8 cytotoxic T cells, through the downregulation of PD-1 expression,41 and the function of natural killer cells42 in mouse models. Taylor et al have recently further shown that the GSK3 inhibitor SB415286 could effectively suppress the growth of mouse EV4 lymphoma xenografts in vivo by reactivation of CD8 T cells.43
To our knowledge, we are the first to comprehensively demonstrate that lymphoma cell lines, as well as primary patient samples, are sensitive to GSK3 inhibition and to describe potential mechanisms of that inhibition. Our data demonstrate the relevance of targeting GSK3, as well as the potential for using the new GSK3 inhibitor 9-ING-41, in lymphoma therapy. The comparison of 9-ING-41 treatment and GSK3 knockout support the notion that 9-ING-41 predominantly targets GSK3. GSK3 was among the first protein serine/threonine kinases discovered44 ; although there are many GSK3 inhibitors available to date,45 none are approved for cancer treatment. Only lithium carbonate is approved by the US Food and Drug Administration for bipolar disorder.46 Because lithium is not specific for, or particularly potent in, inhibiting GSK3 activity, it is unlikely to be an effective antilymphoma drug. Given our data showing the direct role of GSK3 in tumor proliferation and survival, as well as its possible role in rejuvenating tumor-killing T cells and natural killer cells, it appears that GSK3 inhibitors, such as 9-ING-41, may constitute a unique class of drug that could target lymphoma and other cancers through several parallel mechanisms. Our data, along with that from other investigators,9 lay the groundwork for the phase 1 trial of 9-ING-41 (NCT03678883), which has begun.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Jan van Deursen and Kun Ling for expert advice and discussion.
This work was supported by the University of Iowa/Mayo Clinic Lymphoma SPORE (National Institutes of Health [NIH], National Cancer Institute [NCI] grant P50 CA97274), the Mayo Clinic Pancreatic SPORE (NIH NCI grant CA102701), and the Henry J. Predolin Foundation Biobank at the Mayo Clinic. This material is also the result of work supported in part with resources and the use of facilities at the Iowa City VAMC.
The contents of this report do not represent the views of the Veterans Administration or the US Government.
Authorship
Contribution: X.W. and T.E.W. designed the experiments, analyzed and interpreted results, and wrote the manuscript; X.W., M.S., J.L., K.N., L.W., and L.Z. conducted experiments and analyzed data; J.A., Y.L., J.K., and K.W. analyzed data and generated figures; and A.J.N., S.M.A., K.W.P., G.A.B., D.D.B., F.G., and D.M.S. discussed and interpreted results and read and approved the manuscript.
Conflict-of-interest disclosure: F.G. and D.M.S. are employees and stockholders of Actuate Therapeutics Inc. D.D.B. is a member of Actuate Therapeutics Scientific Advisory Board and is a stockholder. The remaining authors declare no competing financial interests.
Correspondence: Thomas E. Witzig, Division of Hematology, Department of Medicine, Mayo Clinic, 200 First St SW, Rochester, MN 55905; e-mail: witzig.thomas@mayo.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal