

Key Points
ABT-199 and DMF synergistically and specifically induce cell death in CTCL cells via cooperative inhibition of Bcl-2 and NF-κB.
In vitro and in vivo data suggest this combination as a promising, effective, and nontoxic novel therapeutic approach in CTCL treatment.

Abstract
Therapeutic options for cutaneous T-cell lymphoma (CTCL) are limited and curative treatment regimens are not available. Thus, new targeted and well-tolerated therapeutic approaches are urgently needed. In this respect, we have recently shown that dimethyl fumerate (DMF) inhibits NF-κB acting as a survival factor in CTCL. Similarly, inhibition of the antiapoptotic protein B-cell lymphoma 2 (Bcl-2) has been shown to induce cell death in CTCL especially when combined with histone deacetylase inhibitors. Therefore, we hypothesized that inhibition of Bcl-2 should potentiate NF-κB inhibition in a novel combination treatment of CTCL. We show that, in vitro, the Bcl-2 inhibitors ABT-199 and ABT-263 induced specific cell death in primary CD4+ cells from CTCL patients as well as in the CTCL cell line SeAx, but not in T cells of healthy donors nor in the CTCL cell line HH, which lacks Bcl-2. Combined treatment with ABT-199 and DMF caused synergistic cell death specifically in CTCL cells engaging 2 independent signaling pathways. To verify these findings in vivo, we performed combined ABT-199 and DMF treatment in a xenograft mouse model for CTCL. The combined treatment effectively reduced tumor growth and increased overall survival via synergistic induction of CTCL cell death and suppression of tumor cell proliferation. Essentially, the combination treatment was superior to ABT-199 monotherapy with respect to both efficacy and tolerability. To sum up, our data provide proof of principle for the therapeutic potential of combining Bcl-2 and NF-κB inhibitors in treating CTCL. Next, this potential should be explored further in a clinical study.
Introduction
Cutaneous T-cell lymphomas (CTCLs) are rare malignancies that primarily manifest in skin.1 No curative therapy is available so far. In addition, frequent relapses and treatment-related side effects complicate therapeutic management. Furthermore, existing therapeutic strategies often result in progressive therapy resistance. Thus, there is an urgent need for novel effective and well-tolerated modalities.2,3 New insight into CTCL pathophysiology has led to a better understanding of disease mechanisms followed by identification of novel therapeutic targets.4-6 In particular, it has been shown that the malignant behavior of CTCL cells is rather caused by cell-death resistance than by hyperproliferation.7,8 As B-cell lymphoma 2 (Bcl-2) activity blocks the intrinsic apoptotic pathway, Bcl-2 has been discussed as a potential factor in the observed cell-death resistance.9-12 Moreover, constitutive activation of the transcription factor NF-κB has been shown to cause cell-death resistance, which may explain treatment failure.13-16 These findings emphasize that overcoming cell-death resistance is crucial for successful targeted CTCL therapy. Hence, the aim of our study was to investigate a combination therapy that inhibits the antiapoptotic protein Bcl-2 and, in addition, sensitizes CTCL cells toward specific cell-death stimuli like the NF-κB inhibitor dimethyl fumarate (DMF). Combination therapies are promising for 2 reasons: first, they synergize in breaking cell-death resistance as they target independent apoptosis-related pathways; second, side effects can be attenuated, lowering the respective doses. To our knowledge, we are the first to provide evidence on the in vivo effects of combination therapies for CTCLs that specifically target different apoptosis-related pathways.
Recently, we identified DMF as a potent NF-κB inhibitor in CTCL that specifically induces apoptosis in malignant CTCL cells sparing other healthy cells and tissues both in vitro and in vivo. We further elucidated mechanisms that suggest DMF can also be effective in other NF-κB–dependent malignancies.17-19 Based on these findings, we are currently performing a clinical phase 2 study on DMF treatment in CTCL.
For Bcl-2, it has been shown that inhibiting this protein can increase the sensitivity of apoptosis-resistant cells toward other cell-death stimuli.20-23 Dysregulation, overexpression, and overactivation of Bcl-2 were found in CTCL cells, suggesting a contribution to cell-death resistance.12,24,25 This makes the combination of Bcl-2 inhibitors with apoptosis-stimulating drugs like DMF a good choice for treatment of CTCL.
Small-molecule inhibitors of Bcl-2 are already used in some hematologic and oncologic diseases, for example, lymphatic and myeloid leukemia, mantle cell lymphoma, or multiple myeloma.26-28 The novel Bcl-2 inhibitor ABT-199 is clinically approved as monotherapy in refractory chronic lymphocytic leukemia (CLL).27 ABT-199 shows rather mild side effects, which makes the substance suitable for CTCL treatment. Indeed, there is first in vitro evidence that Bcl-2 inhibitors can enhance the proapoptotic effects of histone deacetylase (HDAC) inhibitors in CTCL and, thus, Bcl-2 inhibitors may be attractive for combination treatments.20,29
Our results show the proapoptotic effects of Bcl-2 inhibitors on CTCL cells, especially Sézary cells, in vitro and in vivo. In addition, we elucidate the underlying mechanisms of these drugs in CTCL cells and analyze the therapeutic potential of Bcl-2 inhibitors both as a monotherapy and in combination with the NF-κB inhibitor DMF. Essentially, the combination therapy turns out to be highly effective in limiting tumor growth and increasing survival in our xenograft mouse model. At the same time, the tolerability turns out to be better compared with ABT-199 monotreatment. Given the promising preclinical results derived from cell-culture experiments and our xenograft mouse model, an immediate translation into the clinic could open up a successful future therapeutic avenue for CTCL.
Materials and methods
Patients
Seven patients with Sézary syndrome (CTCL stage IV) diagnosed according to World Health Organization–European Organisation for Research and Treatment of Cancer (WHO-EORTC) classification of CTCL and criteria of the International Society for Cutaneous Lymphomas were included in the study. As controls, we investigated blood samples of healthy donors (n = 7) (supplemental Table 1, available on the Blood Web site). Informed consent was obtained from all subjects before inclusion. The study was conducted according to ethical guidelines at our institution and the Helsinki Declaration and was approved by the Ethics Committee II of the University of Heidelberg.
Reagents and T cells
ABT-199, ABT-263, and the mouse antibody against human Bcl-2 were obtained from Santa Cruz Biotechnology. DMF and monomethyl fumarate (MMF) were purchased from Sigma-Aldrich. Caspase 3 and cleaved Caspase 3 antibodies came from Cell Signaling Technology; Ki-67 antibodies were obtained from Novacastra. Primary CD4+ T cells were isolated from heparinized whole blood and treated as indicated.18,19,30 Cell lines were obtained from ATCC (HH, J16) or generously donated by Robert Gniadecki and Vibeke Pless from Bispebjerg Hospital (Copenhagen, Denmark) and Kaltoft from Aarhus University (Denmark) (SeAx) or Eichmüller from DKFZ (Heidelberg, Germany) (MyLa), and cultured as described.18
Cell-death assays, qRT-PCR, western blot
Proximity ligation assay
To determine protein interactions, a proximity ligation assay was performed with Duolink proximity ligation assay (PLA) by Sigma-Aldrich as described previously.17 The rabbit antibody against Bak and the mouse antibody against Bax were obtained from Abcam.
siRNA-mediated knockdown
For the bcl-2 knockdown, the AMAXA Cell Line Nucleofactor kit T (Lonza) was used, and the transfection was performed as described earlier.35 FlexiTube GeneSolution GS596 small-interfering RNAs (siRNAs) for Bcl-2 were obtained from Qiagen.
In vivo xenograft CTCL mouse model and treatment of mice
All animal experiments were in accordance with the approved guidelines of the local Governmental Committee for Animal Experimentation (RP Karlsruhe, Germany, license G282/15). Mice were maintained at a 12-hour light-dark cycle with unrestricted diet and water. Under isoflurane inhalation anesthesia (1% to 1.5% in O2, 0.5 L/min), 3 × 106 SeAx cells suspended in 30 µL of phosphate-buffered saline (PBS)/Matrigel (1:1, vol/vol) were injected intradermally into the right flank of 6- to 7-week-old female NOD SCID γ (NSG) mice recruited from the Center for Preclinical Research (DKFZ, Heidelberg, Germany) and kept on Kliba diet 3307 with n = 8 mice per group. When tumors reached palpable size (mean of 0.025 cm3), mice were randomized and treated first as indicated once by oral gavage with ABT-199 dissolved in a vehicle of 50% propylene glycol, 30% polyethylene glycol 400, and 10% ethanol at a concentration of 100 mg/kg body weight, 10 µL/g mouse, or vehicle alone as indicated. One day later, intraperitoneal treatment with DMF once daily for 28 days was started as indicated. The compound, dissolved at 3 mg/mL in 30°C prewarmed PBS, was given with 10 µL/g mouse at a dose of 30 mg/kg body weight. For control, PBS (10 µL/g mouse) was injected. Tumor volume was measured with a caliper 2 to 3 times a week and calculated according to the formula: V = (length (mm) × width (mm)2)/2. Necropsies were taken when 1 tumor diameter reached 1.5 cm or when mice reached stop criterion of the German Society of Laboratory Animal Sciences (GV-SOLAS), here defined as survival.
For tissue staining, the liver and one-quarter of the tumor was fixed in 4% PBS-buffered formaldehyde and embedded into paraffin according to routine procedures.
Histologic and immunohistochemical stainings
Hematoxylin-and-eosin (H&E) staining as well as immunohistochemistry were performed on 5-µm paraffin sections as described. For immunohistochemical stainings, paraffin-embedded tissue was sectioned into 5-µm slices. Ki-67 was detected in deparaffinized tissue specimens after antigen retrieval (20 minesu at boiling temperature in 10 mM sodium citrate, pH 6.0), block of endogenous peroxidases in 3% H2O2 in PBS for 10 minutes, block in 100% goat serum for 1 hour at room temperature (RT), and incubation overnight at 4°C in a 1/250 dilution of anti–Ki-67 antibodies (NCL-Ki67p; Novacastra). Slides were incubated with appropriate horseradish peroxidase (HRP)-coupled secondary antibody (1/200) for 1 hour at RT, incubated with HRP substrate solution (DAB/H2O2; Sigma, Munich/Germany). Nuclei were counterstained with hematoxylin for 1 minute. Mounting was in Eukitt (R. Langenbrinck, Teningen, Germany). Sections incubated without primary antibody were included as negative controls. Immunohistochemical assessment of the tissue slides was performed by a dermatologist (J.P.N.) and a pathologist (T.A.).
Microscopy
For microscopy, the following Nikon objective lenses were used (Nikon, Tokyo, Japan): Nikon Plan Apoλ 2×/0.1, Nikon Plan Apoλ 20×/0.75. Microscopy was performed at 22°C throughout with cryopreserved or paraffin sections. As fluorochromes phycoerythrin (PE), fluorescein isothiocyanate (FITC), and 4′,6-diamidino-2-phenylindole (DAPI) were used for red, green, and blue stainings, respectively. The micrographs were taken with a Nikon DS-Fi2 camera using the Nikon DS-L3 software.
Statistical analyses
Data are presented as the mean plus or minus standard error of the mean. Two-sided tests were used throughout and the differences were considered statistically significant at P < .05. Pairwise (univariate) comparisons were performed using the Student t test or the Mann-Whitney U test as appropriate. Normalizations were performed as described in the figure legends or supplemental data.
Results
Bcl-2 inhibitors induce cell death in primary patient CTCL cells and CTCL cell lines
We isolated primary CD4+ CTCL cells from the peripheral blood of 7 stage IV CTCL patients and the CD4+ cells from 7 healthy donors and treated them with increasing concentrations of the Bcl-2 inhibitors ABT-199 and ABT-263. Both inhibitors induced a dose-dependent specific CD4+ T-cell death after 24 hours and 48 hours with significantly higher rates in Sézary patients’ cells than in those from healthy donors (Figure 1A-B). This CTCL-specific effect was independent from the tumor burden in blood or skin of the patients or therapeutic regimens applied (supplemental Table 1). To substantiate these results, we also analyzed cell-death induction upon Bcl-2 inhibition in established CTCL cell lines SeAx and MyLa. Both showed high cell-death sensitivity toward Bcl-2 inhibition, whereas the non-CTCL cell line J16 showed a by far attenuated cell-death sensitivity, further corroborating the CTCL-specific effect of Bcl-2 inhibition (Figure 1C-D; supplemental Figure 1). Intriguingly, the CTCL cell line HH showed barely any response to the treatment. In a next step, we set out to correlate the observed cell-death rates with the Bcl-2 expression of the cells. Therefore, we quantified Bcl-2 in the tested cell lines on the messenger RNA (mRNA) level by qRT-PCR as well as on the protein level by western blot analysis. We detected almost no expression of Bcl-2 in the HH cell line in comparison with the high Bcl-2 expression in SeAx cells (Figure 1E-F; supplemental Figure 2). Therefore, the observed CTCL cell-death induction by Bcl-2 inhibitors correlates with Bcl-2 expression of the respective cells. The weak effect in J16 cells and primary healthy CD4+ T cells confirmed that this cell-death effect is specific for malignant CTCL cells and suggested low side effects on benign bystander T cells (Figure 1).
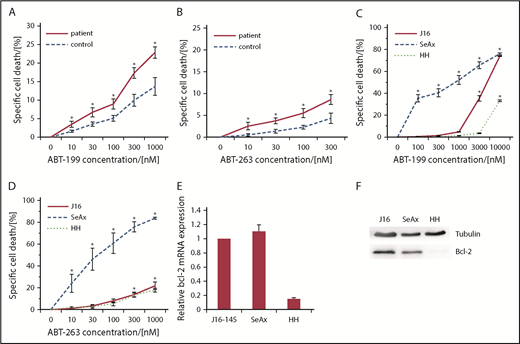
Bcl-2 inhibitors induce cell death in primary patient CTCL cells and CTCL cell lines. (A-B) Specific cell death in primary CD4+ cells isolated from 6 healthy donors (control) and 6 patients with Sézary syndrome (patient) upon treatment with different concentrations of ABT-199 (A) or ABT-263 (B) for 24 hours. *P < .05 from respective healthy controls. (C-D) Specific cell death in J16, HH and SeAx cells upon treatment with different concentrations of ABT-199 (C) or ABT-263 (D) (n = 4, each) for 24 hours. *P < .05 from untreated control. (E) Relative Bcl-2 expression measured by qRTPCR in J16, SeAx and HH cells without stimulation. (F) Western blot analysis of Bcl-2 protein expression in J16, SeAx, and HH cells without stimulation.
Bcl-2 inhibitors induce cell death in primary patient CTCL cells and CTCL cell lines. (A-B) Specific cell death in primary CD4+ cells isolated from 6 healthy donors (control) and 6 patients with Sézary syndrome (patient) upon treatment with different concentrations of ABT-199 (A) or ABT-263 (B) for 24 hours. *P < .05 from respective healthy controls. (C-D) Specific cell death in J16, HH and SeAx cells upon treatment with different concentrations of ABT-199 (C) or ABT-263 (D) (n = 4, each) for 24 hours. *P < .05 from untreated control. (E) Relative Bcl-2 expression measured by qRTPCR in J16, SeAx and HH cells without stimulation. (F) Western blot analysis of Bcl-2 protein expression in J16, SeAx, and HH cells without stimulation.
Bcl-2 inhibitors and DMF synergistically induce cell death in primary patient CTCL cells and CTCL cell lines
In CTCL cells, the thiol-modifying agent and NF-κB inhibitor DMF has been shown to induce apoptosis in CTCL cells in a specific manner.17,18 Therefore, we examined a possible synergistic effect of simultaneous DMF-induced NF-κB inhibition and Bcl-2 inhibition. Indeed, upon combination treatment, we observed a highly significant cooperative effect of the drugs in isolated primary T cells of Sézary patients that by far exceeded the additive values of both monotreatments (Figure 2A left panel; supplemental Figure 3A). Here, both Bcl-2 inhibitors showed similar results. In the CTCL cell line SeAx, we observed a weaker, but still significant, overadditive effect with ABT-199 and DMF (Figure 2A middle panel; supplemental Figure 3B). Combination treatment of HH cells lacking Bcl-2 expression did not have a similar effect, that is, the DMF-induced cell death could not be increased by combination with Bcl-2 inhibitors (Figure 2A right panel; supplemental Figure 3C).
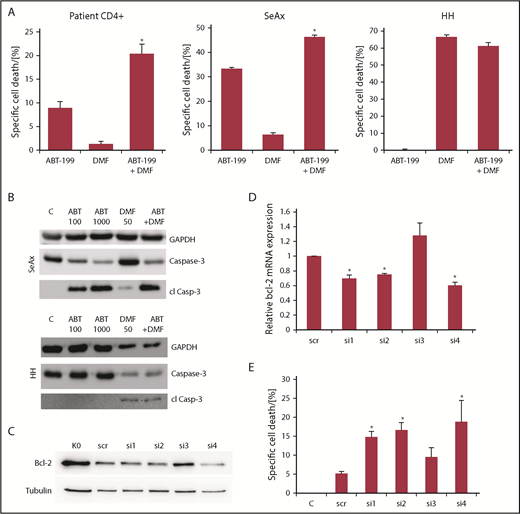
Bcl-2 inhibitors and DMF synergistically induce cell death in primary patient CTCL cells and CTCL cell lines. (A) Specific cell death in primary CD4+ cells isolated from 5 patients with Sézary syndrome (left), SeAx (middle) or HH cells (right) (n = 4 for HH and SeAx) upon treatment with 100 nM ABT-199, 30 µM DMF or the combination of both drugs. *P < .05 from ABT-199 monotreatment. (B) Western blot analysis of caspase 3 cleavage in SeAx and HH cells (as indicated) after treatment with 100 or 1000 nM ABT-199, 50 µM DMF or the combination of 1000 nM ABT-199 and 50µM DMF, as indicated. (C) Representative western blot analysis of SeAx Bcl-2 expression following AMAXA transfection with different siRNAs against bcl-2 after 24 hours. (D) Quantification of signal intensities from western blots for Bcl-2 in SeAx cells following AMAXA transfection with different siRNAs against Bcl-2 after 24 hours (n = 4). *P < .05 from scrambled siRNA control. (E) Specific cell death in SeAx cells transfected with different siRNAs against bcl-2 for 24 hours and treated with 30 µM DMF for another 24 hours (n = 4). *P < .05 from scrambled siRNA control.
Bcl-2 inhibitors and DMF synergistically induce cell death in primary patient CTCL cells and CTCL cell lines. (A) Specific cell death in primary CD4+ cells isolated from 5 patients with Sézary syndrome (left), SeAx (middle) or HH cells (right) (n = 4 for HH and SeAx) upon treatment with 100 nM ABT-199, 30 µM DMF or the combination of both drugs. *P < .05 from ABT-199 monotreatment. (B) Western blot analysis of caspase 3 cleavage in SeAx and HH cells (as indicated) after treatment with 100 or 1000 nM ABT-199, 50 µM DMF or the combination of 1000 nM ABT-199 and 50µM DMF, as indicated. (C) Representative western blot analysis of SeAx Bcl-2 expression following AMAXA transfection with different siRNAs against bcl-2 after 24 hours. (D) Quantification of signal intensities from western blots for Bcl-2 in SeAx cells following AMAXA transfection with different siRNAs against Bcl-2 after 24 hours (n = 4). *P < .05 from scrambled siRNA control. (E) Specific cell death in SeAx cells transfected with different siRNAs against bcl-2 for 24 hours and treated with 30 µM DMF for another 24 hours (n = 4). *P < .05 from scrambled siRNA control.
To confirm the apoptotic feature of the observed cell death by combined NF-κB and Bcl-2 inhibition, we tested caspase cleavage. We found a dose-dependent caspase 3 cleavage in SeAx cells upon treatment with ABT-199 alone or in combination with DMF. This correlated with cell death measured by flow cytometry. In contrast, in HH cells, only DMF could induce significant caspase 3 cleavage. ABT-199 had no effect, most likely due to the lack of Bcl-2 and the missing cell-death induction by ABT-199 (Figure 2B).
To confirm the synergistic effect of decreasing Bcl-2 and NF-κB activity in CTCL cells, we performed a bcl-2 knockdown in SeAx cells. Therefore, the cells were transfected with anti-bcl-2-siRNAs via AMAXA nucleofection. As shown in Figure 2C-D, transfection with different siRNAs caused decreased Bcl-2 expression levels in cells treated with siRNAs 1, 2, and 4 and ranged from 60% to 75% compared with the level of the control cells treated with scrambled siRNA. Twenty-four hours after transfection, we treated the transfected cells with 30 µM DMF for another 24 hours. Cell death upon DMF treatment in this study is illustrated in Figure 2E. We found a direct correlation between Bcl-2 expression and cell-death sensitivity in the transfected cells, that is, lower Bcl-2 activity led to higher cell death. Therefore, we concluded that loss of Bcl-2 sensitizes CTCL cells toward other cell-death stimuli like NF-κB inhibition. This experiment further supported the observed synergistic effect of the combined small-molecule Bcl-2 and NF-κB inhibitors. Mechanistically, the synergistic cell-death induction by combined inhibition of Bcl-2 and NF-κB is not caused by mutual alterations of their signaling cascade, but rather by blocking 2 independent pathways. Upon Bcl-2 knockdown, we found no alterations of the IκBα-mRNA expression on qRT-PCR level (supplemental Figure 4A). IκBα inhibits NF-κB subunits via binding in the cytoplasm, and IκBα expression is used as a readout for activity of the transcription factor NF-κB, as it represents a target gene of NF-κB.18,37,38 This lead to the suggestion that Bcl-2 does not directly regulate NF-κB activity (supplemental Figure 4A). Vice versa, we treated J16, HH, and SeAx cells with DMF and its metabolite MMF and measured the protein levels of Bcl-2 by western blot analysis. Here, no influence on the levels of Bcl-2 or other antiapoptotic Bcl-2 family members like Mcl-1 could be detected in any of the cell lines upon NF-κB inhibition (supplemental Figure 4B). These results strongly suggest that here, the NF-κB and Bcl-2 signaling pathways do not influence or regulate each other directly in their activities.
Bcl-2 inhibition induces Bax/Bak oligomerization in CTCL cell lines depending on their Bcl-2 level
To further elucidate the mechanisms of cell-death induction observed upon ABT-199 and DMF treatment, we used a PLA that indicates the interaction between Bax and Bak at an early step in the signaling cascade leading to apoptosis.10,11 With this method, proximity of the 2 proteins Bax and Bak can be visualized by green fluorescence. If not inhibited, Bcl-2 binds the 2 proteins at the mitochondrial membrane and, thus, prevents them from oligomerization and pore formation. Upon Bcl-2 inhibition, Bcl-2 should release Bax and Bak so that they can form pores through which cytochrome c leaves the mitochondria and completes the apoptosome. Therefore, the detection of green fluorescence confirms effective Bcl-2 inhibition and Bax/Bak interaction. Indeed, we found a significant increase in green fluorescence upon ABT-199 treatment in SeAx cells. This effect was not further enhanced by DMF treatment (Figure 3). Thereby we could confirm the direct effect of Bcl-2 inhibition on apoptosis induction in this cell line. As expected, DMF and ABT-199 had no effect on Bax/Bak interaction in HH cells further confirming our previous findings (supplemental Figure 5).
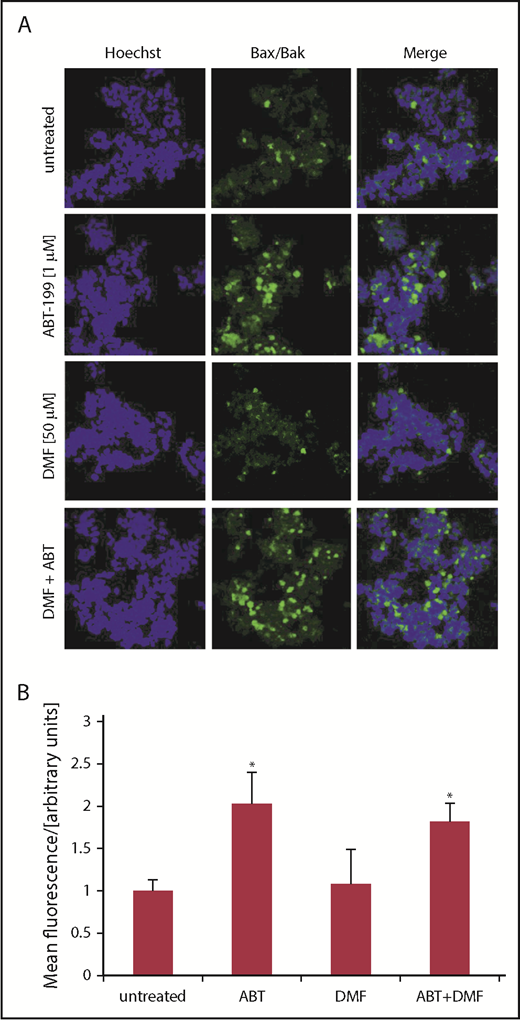
Bcl-2 inhibition induces Bax/Bak oligomerization in CTCL cell lines depending on their Bcl-2 activity. (A) Complex formation of Bax and Bak was determined by PLA in SeAx cells either treated with ABT-199, DMF or the combination of both drugs, as indicated. Vehicle treated cells served as control. Bax/Bak association is depicted by green fluorescence; blue fluorescence indicates nuclear staining with Hoechst dye; shown are representative immunofluorescence analyses from z-stacks. Original magnification ×400. (B) Quantification of mean fluorescence intensities of PLA signals (n = 4). *P < .05 from untreated control.
Bcl-2 inhibition induces Bax/Bak oligomerization in CTCL cell lines depending on their Bcl-2 activity. (A) Complex formation of Bax and Bak was determined by PLA in SeAx cells either treated with ABT-199, DMF or the combination of both drugs, as indicated. Vehicle treated cells served as control. Bax/Bak association is depicted by green fluorescence; blue fluorescence indicates nuclear staining with Hoechst dye; shown are representative immunofluorescence analyses from z-stacks. Original magnification ×400. (B) Quantification of mean fluorescence intensities of PLA signals (n = 4). *P < .05 from untreated control.
Combination of Bcl-2 and NF-κB inhibition decreases tumor growth and increases survival in vivo
To prove the in vivo relevance of our findings, we used a CTCL xenograft mouse model. We intradermally injected the SeAx cell line into NSG mice. After the detection of intradermal tumor growth, the animals were randomized into 4 therapy groups: 1 vehicle-treated control group, 2 monotherapy groups with either ABT-199 or DMF treatment, and a combination therapy group treated with ABT-199 and DMF together (Figure 4). Within the first 10 days after ABT-199 monotreatment, 3 of 8 animals dropped out of this group, whereas in the vehicle group only 1 of 8 mice dropped out within the first 2 weeks of vehicle application. First, we confirmed the xenograft tumors to consist of SeAx cells by immunohistochemistry (supplemental Figure 6). As a primary readout of the therapeutic effect, we assessed growth of the xenograft tumors. Upon treatment, we observed a weak, but not significant, decrease in tumor growth by both DMF and ABT-199 monotreatments. The combination of both drugs, however, led to a significant and lasting stable reduction of tumor growth starting already after 7 days of treatment (Figure 4A). A >40% reduction of SeAx xenograft tumors was seen in the DMF/ABT-199 group at day 18, an effect that could never be reached by single treatment (Figure 4B).18 In accordance with our in vitro experiments, we could confirm an overadditive therapeutic effect of the combination treatment in vivo (Figure 4A-B). Similar synergistic effects of the combination treatment could be observed in the survival rates of the treated animals. The first animals in the PBS control group had to be euthanized due to reaching the end point already after 22 days, whereas this was necessary in the combination treatment group only after 26 days (Figure 4C). Figure 4C shows an overall survival benefit by DMF monotreatment and the combination treatment with DMF and ABT-199. Based on our data, ABT-199 monotreatment shows no advantage with respect to survival (Figure 4C). At day 30, 50% of the animals in the combination treatment group were still under follow-up compared with 13% in the DMF group and 0% in both other groups. This finding emphasizes the beneficial therapeutic effect of combined DMF and ABT-199 therapy (Figure 4D). In summary, we could show a significant increase of the mean survival time by >20% as result of the combination treatment (Figure 4E). Although Bcl-2 inhibition with ABT-199 did not show sufficient therapeutic effects in the monotreatment in vivo, it was able to strikingly reduce tumor growth and to increase survival rates if used in combination treatment (Figure 4). This effect had already been described with different combination partners in other tumor entities.22,23,39 In contrast to the HH mouse model we had established previously to evaluate DMF monotreatment in vivo,18 in the present SeAx xenograft model, no spreading of the malignant T cells to distant organs (liver, spleen, and lymph nodes) was detectable. Therefore, in this mouse model, only the effects of the treatment on the primary xenograft tumors could be evaluated.
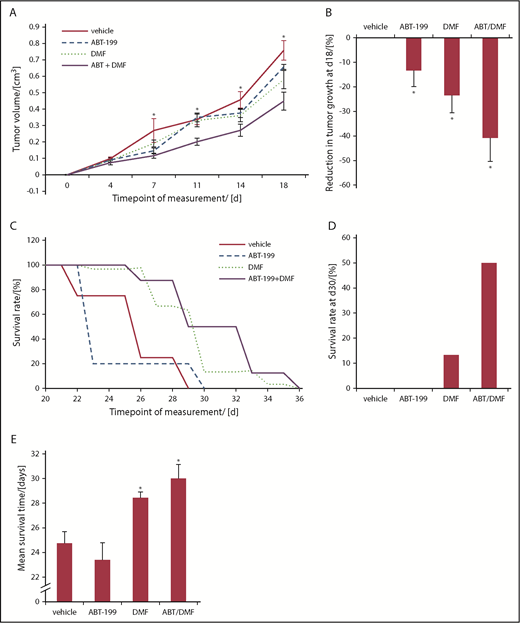
Combined Bcl-2 and NF-κB inhibition inhibits CTCL tumor growth and increases survival in an orthotopic SeAx xenograft model. NSG mice were xenografted with SeAx cells intradermally. Treatment of transplanted mice was with either vehicle, 1 treatment of ABT-199, once daily treatment with DMF for 28 days or the combination of a single application of ABT-199 and the 28 days treatment with a daily application of DMF (n = 8, each). (A) Median tumor volume upon treatment with vehicle, ABT-199, DMF and the combination. *P < .05 of the combination-treatment vs vehicle-treated control. (B) Percent reduction in tumor growth at day 18 of mice treated with PBS, ABT-199, DMF or combination. *P < .05 from vehicle-treated control. (C) Survival curves of xenografted mice in the different treatment groups, as indicated. Decrease in survival is reflecting the reach of the end points defined in "Materials and methods." (D) Percent survival rate of orthotopically xenografted mice treated with vehicle or DMF at day 30, 1 day after the end of the treatment phase. (E) Mean survival time of the SeAx tumor bearers in the different treatment groups, as indicated. *P < .05 vs vehicle-treated control.
Combined Bcl-2 and NF-κB inhibition inhibits CTCL tumor growth and increases survival in an orthotopic SeAx xenograft model. NSG mice were xenografted with SeAx cells intradermally. Treatment of transplanted mice was with either vehicle, 1 treatment of ABT-199, once daily treatment with DMF for 28 days or the combination of a single application of ABT-199 and the 28 days treatment with a daily application of DMF (n = 8, each). (A) Median tumor volume upon treatment with vehicle, ABT-199, DMF and the combination. *P < .05 of the combination-treatment vs vehicle-treated control. (B) Percent reduction in tumor growth at day 18 of mice treated with PBS, ABT-199, DMF or combination. *P < .05 from vehicle-treated control. (C) Survival curves of xenografted mice in the different treatment groups, as indicated. Decrease in survival is reflecting the reach of the end points defined in "Materials and methods." (D) Percent survival rate of orthotopically xenografted mice treated with vehicle or DMF at day 30, 1 day after the end of the treatment phase. (E) Mean survival time of the SeAx tumor bearers in the different treatment groups, as indicated. *P < .05 vs vehicle-treated control.
Combined Bcl-2 and NF-κB inhibition blocks proliferation and induces cell death specifically within CTCL tumors in vivo
After reaching the end points, the tumors were resected for histological analysis of H&E-stained sections. In contrast to the control group, we found significantly larger necrotic tumor areas in the combination group as a sign of tumor cell death and subsequent reduction in tumor size (Figure 5A). Upon quantification with a semiquantitative score, we found a tendency toward increased cell death of the tumor cells in the monotreatment group, compared with the vehicle control. This effect reached statistical significance in the combination group. In addition, we counted the mitosis figures in the H&E specimens and we found a significant decrease in mitotic activity upon treatment with DMF. This effect was even more pronounced upon combination treatment. This finding indicates a deceleration of tumor growth by a reduction of CTCL cell proliferation in the xenograft tumors, especially upon combination treatment (Figure 5C).
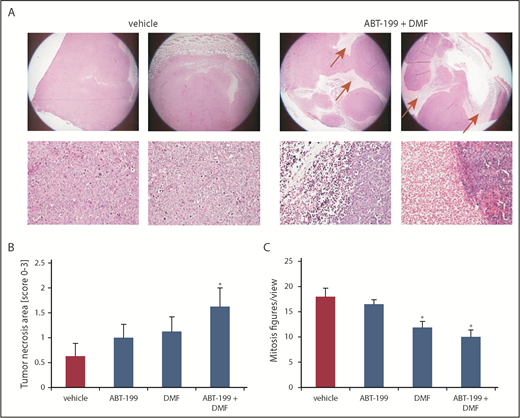
Combined Bcl-2 and NF-κB inhibition HE-morphologically blocks proliferation and induces cell death specifically within CTCL tumors in vivo. SeAx xenograft tumors growing intradermally in NSG mice that were treated as described under Figure 4 were used (A) Representative H&E-stained specimens of primary SeAx tumors from the vehicle control and the combination group (upper panels 20x, lower panels 200x). Red arrows mark necrotic areas. (B) Semiquantitative score of necrotic areas in the primary SeAx xenograft tumors (0% to 25% tumor area covered by necrosis = 0; 25% to 50% tumor area covered by necrosis = 1; 50% to 75% tumor area covered by necrosis = 2; 75% to 100% tumor area covered by necrosis = 3). (C) Quantification of the mitosis count per view under 200x magnification in the 4 treatment groups.
Combined Bcl-2 and NF-κB inhibition HE-morphologically blocks proliferation and induces cell death specifically within CTCL tumors in vivo. SeAx xenograft tumors growing intradermally in NSG mice that were treated as described under Figure 4 were used (A) Representative H&E-stained specimens of primary SeAx tumors from the vehicle control and the combination group (upper panels 20x, lower panels 200x). Red arrows mark necrotic areas. (B) Semiquantitative score of necrotic areas in the primary SeAx xenograft tumors (0% to 25% tumor area covered by necrosis = 0; 25% to 50% tumor area covered by necrosis = 1; 50% to 75% tumor area covered by necrosis = 2; 75% to 100% tumor area covered by necrosis = 3). (C) Quantification of the mitosis count per view under 200x magnification in the 4 treatment groups.
This observation was confirmed by immunohistochemical evaluation of the Ki-67 proliferation index in the tumors. Here, we observed massive proliferative activity in the xenograft tumors of the vehicle controls with up to 33% of the tumor cells positive for Ki-67, especially in the tumor periphery (Figure 6A-B; supplemental Figure 7). The monotreatments with DMF and ABT-199 slightly reduced positivity for Ki-67, whereas in the combination treatment group almost no Ki-67+ cells were detectable (Figure 6B). In a further step, we also stained the tumor sections for cleaved caspase 3 as a readout of apoptotic activity (Figure 6C-D). Indeed, we found slightly increased caspase 3 cleavage within the xenograft tumors upon respective monotreatment with ABT-199 or DMF. However, combination treatment with both drugs resulted in a massive increase in caspase 3 cleavage (Figure 6C-D). In addition, in the combination-treated tumors, we found the cleaved caspase 3+ cells especially close to areas covered by necrosis (Figure 6C; supplemental Figure 8).
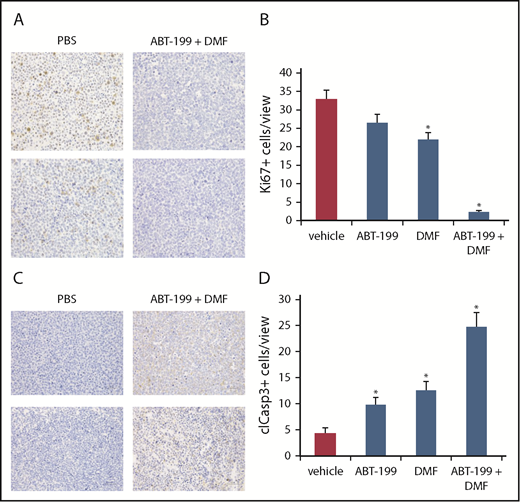
Combined Bcl-2 and NF-κB inhibition blocks proliferation and induces apoptosis specifically within CTCL tumors in vivo in immunohistochemistry. (A) Representative specimens of primary SeAx tumors from the vehicle control and the combination group. Immunohistochemical detection of cellular positivity for the proliferation marker Ki-67 (×400). (B) Quantification of the Ki-67+ cell count per view under ×400 magnification in the 4 treatment groups. (C) Representative specimens of primary SeAx tumors from the vehicle control and the combination group. Immunohistochemical detection of cellular positivity for cleaved caspase-3 as a readout for apoptotic activity (×400). (D) Quantification of the cleaved caspase-3-positive cell count per view under ×400 magnification in the 4 treatment groups.
Combined Bcl-2 and NF-κB inhibition blocks proliferation and induces apoptosis specifically within CTCL tumors in vivo in immunohistochemistry. (A) Representative specimens of primary SeAx tumors from the vehicle control and the combination group. Immunohistochemical detection of cellular positivity for the proliferation marker Ki-67 (×400). (B) Quantification of the Ki-67+ cell count per view under ×400 magnification in the 4 treatment groups. (C) Representative specimens of primary SeAx tumors from the vehicle control and the combination group. Immunohistochemical detection of cellular positivity for cleaved caspase-3 as a readout for apoptotic activity (×400). (D) Quantification of the cleaved caspase-3-positive cell count per view under ×400 magnification in the 4 treatment groups.
Thus, the therapeutic effect of combined Bcl-2 and NF-κB treatment in vivo relies on both cell-death induction and inhibition of proliferation of the CTCL tumor cells.
Discussion
Resistance to distinct cell-death pathways is an important driver for malignant potential of CTCL. Hence, restoring the sensitivity toward apoptosis has become an attractive therapeutic aim in developing novel treatment options. In recent years, growing insight into molecular and cellular mechanisms of CTCL cell biology put forward several potential avenues for targeted CTCL therapy.40‐44 So far, however, only 2 targeted monotherapies, but no targeted combination treatments, are approved based on phase 3 studies: brentuximab-vedotin and mogamulizumab.43,45
Recent studies identified several targets like MAPKs or the transcription factor NF-κB and their related signaling pathways as important drivers of cell-death resistance in CTCL.6,17‐19,46,47 In addition, these studies show that inhibitors of these targets represent potent cell-death stimuli in CTCL.18,19,47 Several studies suggest an important role of Bcl-2 in the observed resistance of CTCL cells toward cell-death stimuli. Bcl-2 acts as an important cell-death blocker in apoptosis.10‐12,48 Indeed, evidence shows that CTCL cells overexpress Bcl-2.9,24
The hypothesis of our study was that the combination of cell-death stimuli with Bcl-2 inhibition can enhance CTCL cell death synergistically. This rationale for our study is underlined by recent in vitro data that show Bcl-2 inhibition can enhance the proapoptotic effect of HDAC inhibitors. Thus, it represents a promising target for combination therapy.20,29 Our present study is the first to investigate the effects of combination treatment with another specific targeted therapy that restores apoptosis sensitivity in CTCL cells, especially Sézary cells. In addition, it investigates and elucidates underlying mechanisms of Bcl-2 inhibition alone and in combination treatment. Furthermore, our study provides first evidence for the in vivo relevance of Bcl-2 inhibition, especially in combination with NF-κB inhibition, in CTCL. The SeAx xenograft mouse model we used for that purpose was different from the models described in the literature so far in some aspects: in contrast to other groups working on SeAx xenografts,49‐51 we intradermally injected a high number of cells at a high density (1 × 108/mL intradermally) in the presence of Matrigel into NSG mice from different sources and, thus, achieved stable and significant xenograft tumor growth. Of note, only SeAx cell batches with basal apoptotic levels of <5% to 10% were inoculated.
Our results show that Bcl-2 inhibition alone already induces a significant amount of apoptosis in CTCL cells. Healthy bystander T cells are far less affected. This finding suggests that in contrast to healthy T cells, CTCL cells show an addiction on constitutive Bcl-2 activity for their survival. The preclinical outcome of CTCL-specific cell death promises a favorable side-effect profile in CTCL patients. The fact that benign bystander T cells might be largely spared especially should not lead to additional immunosuppression. This is highly relevant as immunosuppression and subsequent infections are often limiting the prognosis of CTCL patients.52‐54 Due to the approval of ABT-199 for special hematologic malignancies and several ongoing clinical trials, there are already initial data on the tolerability of ABT-199.55,56 Indeed, the side effects of ABT-199 seem to be rather mild. So far, according to recent clinical studies, they mainly include diarrhea (52%), respiratory infections (48%), nausea (47%), and neutropenia (41%).27,28,57,58 In our xenograft mouse model, however, the drug led to increased morbidity in the ABT-199 monotreatment group. This effect was not observed in the combination treatment group with ABT-199 and DMF, further favoring Bcl-2 inhibitors in combination treatment over monotreatment.
DMF has been found to act as a potent cell-death stimulus in CTCL by blocking NF-κB as an important survival factor of T cells. Therefore, we chose DMF in a combination with Bcl-2 inhibition. We found a strong synergistic effect on cell death by combining DMF and Bcl-2 inhibition in isolated patient T cells. The synergistic effect is due to the fact that DMF and ABT target 2 independent pathways. Consequently, Bcl-2 activity and NF-κB activation can, at least partly, mutually replace the function of each other with respect to protection from apoptosis stimuli. Indeed, there is evidence that NF-κB and Bcl-2 can influence each other’s activity in different conditions of infection and malignancy.59,60 Our data indicate that this effect, if relevant for CTCL, is not exerted by mutual regulation of Bcl-2 and NF-κB on the gene-expression level. Our in vivo data show several important aspects of combined Bcl-2 and NF-κB inhibition: first, the effect of Bcl-2 inhibition monotherapy is by far weaker than in vitro. This typically relies on pharmacokinetics like first-pass effect or increased degradation. In addition, this treatment was given just once during the whole experiment, which might provide a further explanation. Nevertheless, the combinatory effect of dual inhibition is unaffected, independent of this and comparably strong as in vitro. Second, our in vivo data indicate that the combination treatment not only improves the efficacy of the treatment in a synergistic manner but also reduces the necessary dosage and associated side effects, which further supports our rationale to combine inhibition of Bcl-2 and NF-κB.
In summary, we put forward Bcl-2 inhibition in combination with the NF-κB inhibitor DMF as a promising new therapeutic strategy in CTCL. The synergistic proapoptotic effect of this combination on CTCL apoptosis in vitro was confirmed in vivo in a xenograft mouse model. The combination treatment led to reduced CTCL tumor growth and increased mouse survival by inducing CTCL cell death and in addition interfered with CTCL cell proliferation. Importantly, this effect is highly specific on CTCL cells. This promises a good tolerability of this treatment as is observed in our mouse model. With regard to both efficacy and tolerability, the combination therapy turned out to be superior to ABT monotreatment. For these reasons, the presented data suggest this combination therapy to be clinically evaluated in CTCL patients in a clinical trial.
In this project, additional data that could be stored in a public deposit was not generated. Nevertheless, all inquiries concerning sharing data or reagents can be emailed to the corresponding author
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Hiltrud Schönhaber, Diana Vobis, Jochen Weber, Brigitte Steinbauer, Andrea-Pohl Arnold, and Stephanie Laier for excellent technical support. In addition, the authors thank Sven Schneider and the National Center for Tumor Diseases Heidelberg (NCT) Tissue Bank Heidelberg for support with the cell line characterization.
This work was supported by the Deutsche Forschungsgemeinschaft (NI1407/1-2) and the Wilhelm-Sander-Stiftung (project no. 2012.077.1/2.).
Authorship
Contribution: J.P.N. designed the study and planned the experiments; T.C.F., K.M.-D., J.D.B., A.S., and J.P.N. performed experiments; T.C.F., K.M.-D., J.D.B., T.A., A.S., K.G., S.G., P.H.K., and J.P.N. evaluated clinical and experimental data; and T.C.F., K.G., and J.P.N. prepared and wrote the manuscript.
Conflict-of-interest disclosure: J.P.N. received travel and congress participation funding by TEVA and Novartis as well as consulting fees from TEVA, Almirall, Biogen, Novartis, Kyowa Hakko Kirin, Innate Pharma, Takeda, and Actelion. P.H.K. and K.G. received consulting fees from Biogen. The remaining authors declare no competing financial interests.
Correspondence: Jan P. Nicolay, Department of Dermatology, University Medical Center Mannheim, Haus 27, Theodor-Kutzer-Ufer 1-3, 68167 Mannheim, Germany; e-mail: jan.nicolay@umm.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal