

Key Points
Outcomes in a multicenter BPDCN population in the modern era provide a benchmark before targeted therapy.
Age <60 years, normal karyotype, and TdT positivity were associated with improved survival; pralatrexate and enasidenib had activity in BPDCN.

Abstract
Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is an uncommon hematologic malignancy with poor outcomes. Existing data on the clinical behavior of BPDCN are limited because reported outcomes are from small retrospective series, and standardized treatment guidelines are lacking. The interleukin-3 cytotoxin conjugate tagraxofusp was recently tested in phase 1/2 trials that led to US Food and Drug Administration approval, the first ever for BPDCN. However, because there was no matched internal comparator in this or any clinical study to date, results of BPDCN trials testing new drugs are difficult to compare with alternative therapies. We therefore sought to define the clinical characteristics and outcomes of a group of patients with BPDCN treated at 3 US cancer centers in the modern era but before tagraxofusp was available. In 59 studied patients with BPDCN, the median overall survival from diagnosis was 24 months, and outcomes were similar in patients with “skin only” or with systemic disease at presentation. Intensive first-line therapy and “lymphoid-type” chemotherapy regimens were associated with better outcomes. Only 55% of patients received intensive chemotherapy, and 42% of patients underwent stem cell transplantation. Clinical characteristics at diagnosis associated with poorer outcomes included age >60 years, abnormal karyotype, and terminal deoxynucleotidyltransferase (TdT) negativity in the BPDCN cells. We also identified disease responses to pralatrexate and enasidenib in some patients. This study highlights poor outcomes for patients with BPDCN in the modern era and the need for new treatments. Outcomes from ongoing clinical trials for BPDCN can be evaluated relative to this contemporary cohort.
Introduction
Blastic plasmacytoid dendritic cell neoplasm (BPDCN), formerly known by other terms, including agranular CD4+/CD56+ hematodermic neoplasm or blastic natural killer cell lymphoma, is a rare and highly aggressive hematologic malignancy that involves skin, bone marrow, lymph nodes, and/or extranodal sites. Previously believed to originate from natural killer cells, T cells, or monocytes, it was more recently defined as a clonal transformation of plasmacytoid dendritic cells or their precursors.1-3 Although the entity was first described in 1994,4 it has undergone multiple nomenclature revisions. It was recognized in the 2008 World Health Organization (WHO) classification as a subset of acute myeloid leukemia and related neoplasms.5 In the 2016 revision to the WHO taxonomy, BPDCN received its own classification as a distinct hematologic malignancy.6
Despite advances in the diagnosis and understanding of the cell of origin and biology,3,7-10 development of therapies for BPDCN has been slow until recently.11 Currently, there are no consensus guidelines for treatment, but several novel therapies are under investigation.12 Historically, BPDCN treatments were based on chemotherapy regimens for lymphoma, acute lymphoblastic leukemia, or acute myeloid leukemia. Although a number of combination chemotherapy regimens may produce high rates of initial response, they are associated with rapid relapses and a dismal median survival. Hematopoietic stem cell transplantation (HSCT) is the only therapeutic modality that results in durable remissions.13-16
The median age at diagnosis of BPDCN is 53 years, with a bimodal incidence pattern with peaks <20 and >60 years of age; however, the majority of diagnosed patients are >60 years of age.17,18 Older patients with comorbidities may not be suitable candidates for intensive induction chemotherapy or stem cell transplantation. Therefore, novel therapies are being pursued, most of which are repurposed from investigational agents used in other indications such as acute myeloid leukemia. The first prospective clinical trial specifically for BPDCN was conducted in a multicenter study of tagraxofusp-erzs (SL-401; Stemline Therapeutics, Inc.), a recombinant protein composed of a truncated diphtheria toxin payload fused to interleukin-3.19 Tagraxofusp was approved for use in patients with BPDCN in December 2018. However, because of the rarity of BPDCN, trials studying tagraxofusp and other novel therapies are single-arm studies. This makes it challenging to compare BPDCN clinical trial results vs conventional chemotherapy in otherwise matched populations in the contemporary era.
We therefore set out to investigate the outcomes of patients with BPDCN treated with “standard of care” approaches at 3 major referral cancer centers in the United States over the last 17 years. Our goal was to define factors associated with survival, understand therapies used outside of clinical trials, and establish a benchmark of pretargeted therapy outcomes in BPDCN.
Methods
Subjects
This study was approved by the institutional review board at each participating site. We identified all patients diagnosed with BPDCN at the Memorial Sloan Kettering Cancer Center, Massachusetts General Hospital, and Dana-Farber Cancer Institute/Brigham and Women’s Hospital between 2000 and 2017. Patients were identified through an inquiry of the electronic medical record for all patients with a listed diagnosis or pathology report containing the following terms: “blastic plasmacytoid dendritic cell neoplasm” and/or “BPDCN”; ”blastic” AND “NK” AND “lymphoma”; “NK blastic lymphoma”; and “CD4+” AND “CD56+” AND “hematodermic neoplasm.” Data from cases before publication of the WHO definition of BPDCN in 2008 were reviewed by a hematopathologist to ensure they met the modern diagnostic criteria. Data were collected regarding patients’ age at diagnosis, date of diagnosis, staging, initial laboratory and pathologic evaluation (including immunohistochemistry and standard karyotyping on bone marrow or skin tumors), involved sites of disease, previous therapies, response to therapy, duration of response, last follow-up, and date of death. Treatments were categorized as intensive if they generally caused a blood count nadir significant enough to require hospitalization, moderate if they affected blood counts but not necessarily requiring hospitalization, or nonintensive if they were typically given as outpatient and had minimal effect on blood counts. Responses were determined according to the treating physician’s notes and corroborated by contemporaneous results of bone marrow and skin biopsies, dermatologic evaluations, and positron emission tomography and/or computed tomography scans.
Statistical analysis
Fisher’s exact test was used for categorical comparisons. For comparisons of continuous measures between groups, a Wilcoxon rank sum test was used; for the 3-group comparison, a Kruskal-Wallis test was performed. Overall survival (OS) was calculated from the date of diagnosis to the date of death or censored at the date last known alive. For the subset of patients who underwent HSCT, including allogeneic and autologous transplant, survival was also calculated from the date of transplant to the date of death or last known alive. Progression-free survival (PFS) was calculated from the date of initiation of treatment until the date of relapse or death and was also censored at the date last known alive and without progression. PFS for the subset of patients who underwent transplant was also calculated as the time of transplant to the date of death, relapse, or last known alive. OS and PFS were compared between groups by using a log-rank test and estimated by using the Kaplan-Meier method. The cumulative incidences of acute and chronic graft-versus-host disease (GVHD) were calculated from the date of transplant to the time of the event, with death and relapse as competing risks; they were censored at the last known time alive and compared by using the Gray test. All P values reported are 2-sided and considered significant if <.05.
Results
Patient characteristics
A total of 59 patients with BPDCN were diagnosed from 2000 to 2017 at 3 institutions (Memorial Sloan Kettering Cancer Center, Massachusetts General Hospital, and Dana-Farber Cancer Institute/Brigham and Women’s Hospital) and were included in the analysis. The distribution of patients according to year is listed in supplemental Table 1 (available on the Blood Web site). Patient characteristics are included in Table 1 and are stratified according to the extent of organ involvement at diagnosis (11 were “skin only,” with disease involvement limited to skin and specifically not in marrow, blood, or nodes; 10 were “systemic only without skin,” having marrow and typically other organ involvement but without skin involvement; 28 had both skin and systemic involvement); 10 patients had incomplete staging information. The most common site of involvement was skin (present in 80%, or 39 of 49 patients who had complete staging); other frequent sites of involvement included the bone marrow (61%), lymph nodes (43%), and spleen (16%). Rarely, bone, liver, nasopharynx, or central nervous system involvement was seen, as were single cases each of cervix, rectum, or ocular involvement. Certain other characteristics were consistent with what has previously been reported in BPDCN, including an older population (median age at diagnosis, 66 years) and male predominance (4:1 male:female). We also observed the previously reported association with another known malignancy, including other myeloid neoplasms,20-22 either concurrently or according to history in 39% of our cohort.
Patient characteristics according to organ involvement at Dx
| Characteristic | Total | Skin only | Systemic only without skin | Skin and systemic | Incomplete staging | P* |
|---|---|---|---|---|---|---|
| N | 59 | 11 | 10 | 28 | 10 | |
| Age at Dx, median (range), y | 66 (6, 91) | 66 (11, 78) | 61 (26, 84) | 63 (15, 84) | 77 (6, 91) | .98 |
| <60 | 12 (20) | 3 (27) | 4 (40) | 9 (32) | 1 (10) | .84 |
| ≥60 | 47 (80) | 8 (73) | 6 (60) | 19 (68) | 9 (90) | |
| Sex | ||||||
| Female | 12 (20) | 3 (27) | 2 (20) | 5 (18) | 2 (20) | .89 |
| Male | 47 (80) | 8 (73) | 8 (80) | 23 (82) | 8 (80) | |
| Ethnicity | ||||||
| White | 37 (63) | 6 (54) | 6 (60) | 22 (79) | 3 (30) | .20 |
| Other | 9 (15) | 4 (36) | 2 (20) | 3 (10) | 0 (0) | |
| Unknown | 13 (22) | 1 (9) | 2 (20) | 3 (11) | 7 (70) | |
| Karyotype† | ||||||
| Normal | 24 (65) | 9 (100) | 5 (50) | 10 (59) | 0 (0) | .036 |
| Abnormal | 13 (35) | 0 (0) | 5 (50) | 7 (41) | 1 (100) | — |
| Unknown | 22 | 2 | 0 | 11 | 9 | — |
| Other malignancy | ||||||
| Yes | 23 (39) | 3 (27) | 4 (40) | 15 (54) | 1 (10) | .17 |
| None | 15 (25) | 6 (55) | 2 (20) | 6 (21) | 1 (10) | |
| Unknown | 21 (36) | 2 (18) | 4 (40) | 7 (25) | 8 (80) | |
| WBC at Dx, median (range) | 5.3 (1.1, 170.0) | 5.4 (2.6, 10.0) | 5.5 (1.1, 170.0) | 5.2 (1.6, 10.0) | 3.1 | .85 |
| Platelet at Dx, median (range) | 129 (16, 399) | 149 (76, 279) | 90 (29, 399) | 128 (16, 280) | 214 | .41 |
| Hgb at Dx, median (range) | 12.3 (6.9, 16.3) | 13.3 (11.4, 16.3) | 9.7 (6.9, 14.2) | 11.9 (7.6, 16.0) | 13.1 | .019 |
| MCV at Dx, median (range) | 90 (73, 225) | 91.1 (82.0, 111.0) | 89.4 (75.0, 225.0) | 90.0 (73.0, 110.2) | 91.8 | .82 |
| No. of sites, median (range) | 2 (1, 4) | — | — | — | — | — |
| Site of involvement†,‡ | ||||||
| Skin | 39 (80) | 11 (100) | 0 (0) | 28 (100) | 5 (50) | — |
| Bone marrow | 30 (61) | 0 (0) | 10 (100) | 20 (71) | 2 (20) | |
| Lymph node(s) | 21 (43) | 0 (0) | 4 (40) | 17 (61) | 0 (0) | |
| Bone | 1 (2) | 0 (0) | 0 (0) | 1 (4) | 1 (10) | |
| Liver | 1 (2) | 0 (0) | 0 (0) | 1 (4) | 0 (0) | |
| Spleen | 8 (16) | 0 (0) | 2 (20) | 6 (21) | 0 (0) | |
| CNS | 3 (6) | 0 (0) | 1 (10) | 2 (7) | 0 (0) | |
| Other§ | 5 (10) | 0 (0) | 1 (10) | 4 (14) | 1 (10) |
| Characteristic | Total | Skin only | Systemic only without skin | Skin and systemic | Incomplete staging | P* |
|---|---|---|---|---|---|---|
| N | 59 | 11 | 10 | 28 | 10 | |
| Age at Dx, median (range), y | 66 (6, 91) | 66 (11, 78) | 61 (26, 84) | 63 (15, 84) | 77 (6, 91) | .98 |
| <60 | 12 (20) | 3 (27) | 4 (40) | 9 (32) | 1 (10) | .84 |
| ≥60 | 47 (80) | 8 (73) | 6 (60) | 19 (68) | 9 (90) | |
| Sex | ||||||
| Female | 12 (20) | 3 (27) | 2 (20) | 5 (18) | 2 (20) | .89 |
| Male | 47 (80) | 8 (73) | 8 (80) | 23 (82) | 8 (80) | |
| Ethnicity | ||||||
| White | 37 (63) | 6 (54) | 6 (60) | 22 (79) | 3 (30) | .20 |
| Other | 9 (15) | 4 (36) | 2 (20) | 3 (10) | 0 (0) | |
| Unknown | 13 (22) | 1 (9) | 2 (20) | 3 (11) | 7 (70) | |
| Karyotype† | ||||||
| Normal | 24 (65) | 9 (100) | 5 (50) | 10 (59) | 0 (0) | .036 |
| Abnormal | 13 (35) | 0 (0) | 5 (50) | 7 (41) | 1 (100) | — |
| Unknown | 22 | 2 | 0 | 11 | 9 | — |
| Other malignancy | ||||||
| Yes | 23 (39) | 3 (27) | 4 (40) | 15 (54) | 1 (10) | .17 |
| None | 15 (25) | 6 (55) | 2 (20) | 6 (21) | 1 (10) | |
| Unknown | 21 (36) | 2 (18) | 4 (40) | 7 (25) | 8 (80) | |
| WBC at Dx, median (range) | 5.3 (1.1, 170.0) | 5.4 (2.6, 10.0) | 5.5 (1.1, 170.0) | 5.2 (1.6, 10.0) | 3.1 | .85 |
| Platelet at Dx, median (range) | 129 (16, 399) | 149 (76, 279) | 90 (29, 399) | 128 (16, 280) | 214 | .41 |
| Hgb at Dx, median (range) | 12.3 (6.9, 16.3) | 13.3 (11.4, 16.3) | 9.7 (6.9, 14.2) | 11.9 (7.6, 16.0) | 13.1 | .019 |
| MCV at Dx, median (range) | 90 (73, 225) | 91.1 (82.0, 111.0) | 89.4 (75.0, 225.0) | 90.0 (73.0, 110.2) | 91.8 | .82 |
| No. of sites, median (range) | 2 (1, 4) | — | — | — | — | — |
| Site of involvement†,‡ | ||||||
| Skin | 39 (80) | 11 (100) | 0 (0) | 28 (100) | 5 (50) | — |
| Bone marrow | 30 (61) | 0 (0) | 10 (100) | 20 (71) | 2 (20) | |
| Lymph node(s) | 21 (43) | 0 (0) | 4 (40) | 17 (61) | 0 (0) | |
| Bone | 1 (2) | 0 (0) | 0 (0) | 1 (4) | 1 (10) | |
| Liver | 1 (2) | 0 (0) | 0 (0) | 1 (4) | 0 (0) | |
| Spleen | 8 (16) | 0 (0) | 2 (20) | 6 (21) | 0 (0) | |
| CNS | 3 (6) | 0 (0) | 1 (10) | 2 (7) | 0 (0) | |
| Other§ | 5 (10) | 0 (0) | 1 (10) | 4 (14) | 1 (10) |
Data are presented as no. (%) unless otherwise indicated. Dx, diagnosis; Hgb, hemoglobin; MCV, mean corpuscular volume; WBC, white blood cell.
Test excludes patients in the Incomplete staging column, who had incomplete data on sites of involvement.
Percentage in Total column excludes patients with incomplete staging.
Multiple sites possible per patient.
Three nasopharynx, 1 cervix, 1 ocular, and 1 rectum.
Of the 59 patients, 37 had cytogenetic analysis performed; 24 (65%) of 37 had a normal karyotype; 13 (35%) of 37 exhibited an abnormal karyotype, of which 11 (85%) were complex, defined as ≥3 abnormalities (supplemental Table 2). Of patients with skin-only initial presentations, 9 had cytogenetic analysis, all of which exhibited a normal karyotype; 2 patients did not have karyotypic information available. In contrast, among those with both skin and systemic involvement, 59% (10 of 17) had normal karyotype and 41% (7 of 17) had an abnormal karyotype (Fisher exact test for skin only vs both skin and systemic, P = .036). One case had evidence of an MYC gene rearrangement, as has been reported previously in patients with BPDCN in association with poorer prognosis.7,10,23 Pathologic markers tested by using immunohistochemistry and/or flow cytometry were consistent with those previously reported for BPDCN, including positivity for CD123, CD4, and CD56 in the majority of cases (supplemental Table 3).
Patients in this cohort received at least 22 different frontline treatment regimens, highlighting the heterogeneity of BPDCN treatment even in the modern era (Table 2; supplemental Table 4). With so many different treatments, it was difficult to draw any conclusions about the effectiveness of individual therapies; we therefore combined them into different categories according to intensity and whether the regimen was typically used to treat lymphoid or myeloid malignancies. Only 55% received intensive induction-type chemotherapy, which was the recommended approach for younger, fit patients before tagraxofusp approval.11 The remaining 45% received either moderate-intensity (25%) or nonintensive (20%) therapy. The majority of patients received lymphoid-type regimens, whereas some received myeloid-type regimens. All patients with skin-only involvement received a lymphoid-type regimen compared with 80% of those with skin and systemic involvement and 50% of those with systemic disease but no skin involvement (P = .039). Patients with skin-only disease also had a longer median time from diagnosis to first treatment (42 days) than those with skin and systemic (23 days) or systemic without skin (12 days) involvement (P = .048).
Treatment summary
| Variable | Total | Skin only | Systemic only without skin | Skin and systemic | Incomplete staging | P* |
|---|---|---|---|---|---|---|
| N | 44 | 10 | 8 | 25 | 1 | |
| Time from Dx to first treatment, median (range), d | 27 (2, 164) | 42 (5, 164) | 12 (3, 40) | 23 (2, 101) | 56 | .048 |
| No. of therapies, median (range)* | 2 (1, 6) | 2 (1, 5) | 2 (1, 6) | 2 (1, 6) | 2 | .78 |
| First treatment† | ||||||
| HyperCVAD ± other | 8 (18) | 1 (10) | 2 (25) | 5 (20) | 0 (0) | — |
| ICE | 6 (14) | 2 (20) | 0 (0) | 4 (16) | 0 (0) | |
| CALGB 9111 | 5 (11) | 1 (10) | 2 (25) | 2 (8) | 0 (0) | |
| 7+3 ± other | 5 (11) | 0 (0) | 2 (25) | 3 (12) | 0 (0) | |
| CHOP ± other | 5 (11) | 2 (20) | 0 (0) | 2 (8) | 1 (100) | |
| Pralatrexate | 3 (7) | 0 (0) | 0 (0) | 3 (8) | 0 (0) | |
| HiDAC ± other | 2 (5) | 0 (0) | 1 (12) | 1 (4) | 0 (0) | |
| Gemcitabine based | 2 (5) | 1 (10) | 0 (0) | 1 (4) | 0 (0) | |
| Intensive ALL | 2 (5) | 1 (10) | 0 (0) | 1 (4) | 0 (0) | |
| Other | 6 (14) | 2 (20) | 1 (12) | 3 (12) | 0 (0) | |
| Type of first therapy | ||||||
| Lymphoid | 35 (77) | 10 (100) | 4 (50) | 20 (80) | 1 (100) | .039 |
| Myeloid | 9 (21) | 0 (0) | 4 (50) | 5 (20) | 0 (0) | |
| Intensity of first therapy | ||||||
| Not intensive | 9 (20) | 2 (20) | 2 (25) | 5 (20) | 0 (0) | .54 |
| Moderate | 11 (25) | 3 (30) | 0 (0) | 7 (28) | 1 (100) | |
| Intensive | 24 (55) | 5 (50) | 6 (75) | 13 (52) | 0 (0) |
| Variable | Total | Skin only | Systemic only without skin | Skin and systemic | Incomplete staging | P* |
|---|---|---|---|---|---|---|
| N | 44 | 10 | 8 | 25 | 1 | |
| Time from Dx to first treatment, median (range), d | 27 (2, 164) | 42 (5, 164) | 12 (3, 40) | 23 (2, 101) | 56 | .048 |
| No. of therapies, median (range)* | 2 (1, 6) | 2 (1, 5) | 2 (1, 6) | 2 (1, 6) | 2 | .78 |
| First treatment† | ||||||
| HyperCVAD ± other | 8 (18) | 1 (10) | 2 (25) | 5 (20) | 0 (0) | — |
| ICE | 6 (14) | 2 (20) | 0 (0) | 4 (16) | 0 (0) | |
| CALGB 9111 | 5 (11) | 1 (10) | 2 (25) | 2 (8) | 0 (0) | |
| 7+3 ± other | 5 (11) | 0 (0) | 2 (25) | 3 (12) | 0 (0) | |
| CHOP ± other | 5 (11) | 2 (20) | 0 (0) | 2 (8) | 1 (100) | |
| Pralatrexate | 3 (7) | 0 (0) | 0 (0) | 3 (8) | 0 (0) | |
| HiDAC ± other | 2 (5) | 0 (0) | 1 (12) | 1 (4) | 0 (0) | |
| Gemcitabine based | 2 (5) | 1 (10) | 0 (0) | 1 (4) | 0 (0) | |
| Intensive ALL | 2 (5) | 1 (10) | 0 (0) | 1 (4) | 0 (0) | |
| Other | 6 (14) | 2 (20) | 1 (12) | 3 (12) | 0 (0) | |
| Type of first therapy | ||||||
| Lymphoid | 35 (77) | 10 (100) | 4 (50) | 20 (80) | 1 (100) | .039 |
| Myeloid | 9 (21) | 0 (0) | 4 (50) | 5 (20) | 0 (0) | |
| Intensity of first therapy | ||||||
| Not intensive | 9 (20) | 2 (20) | 2 (25) | 5 (20) | 0 (0) | .54 |
| Moderate | 11 (25) | 3 (30) | 0 (0) | 7 (28) | 1 (100) | |
| Intensive | 24 (55) | 5 (50) | 6 (75) | 13 (52) | 0 (0) |
Test excludes patients in the Incomplete staging column.
HyperCVAD, hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone; ICE, ifosfamide, carboplatin, etoposide; 7+3, cytarabine, daunorubicin (or idarubicin); CALGB 9111, induction cyclophosphamide, daunorubicin, vincristine, prednisolone, asparaginase, and filgrastim; CHOP, cyclophosphamide, doxorubicin, vincristine, prednisolone; HIDAC, high-dose cytarabine; Intensive ALL, regimens used for high-risk pediatric acute lymphoblastic leukemia or Burkitt lymphoma (eg, SMILE [dexamethasone, methotrexate, ifosfamide, l-asparaginase, etoposide]); Other, targeted therapy or radiation.
Hematopoietic stem cell transplantation
Approximately 42% of patients (25 of 59) underwent HSCT at any time during their disease course (average, 182 days after diagnosis); all underwent transplant while in remission (Table 3). Five of the 25 received high-dose chemotherapy and autologous stem cell transplantation. Twenty of 25 underwent allogeneic HSCT, with most receiving peripheral blood–mobilized stem cells (n = 18) as the graft source. Nine (45%) allogeneic transplants used myeloablative conditioning regimens and 11 (55%) used reduced intensity conditioning regimens. The cumulative incidences of grade 2 to 4 acute GVHD and any grade chronic GVHD after allogeneic transplant were 42% (95% confidence interval [CI], 20-63) and 20% (95% CI, 6-40), respectively (supplemental Figure 1). Given the sample size, as well as the heterogeneous conditioning regimens and GVHD prophylaxis regimens used, this data set is not powered to explore transplant toxicities in specific subsets. However, we saw no obvious increase in GVHD of the skin or other sites among patients with or without BPDCN involvement of the skin at the time of diagnosis (Table 4).
Transplant characteristics
| Characteristic | Total | Skin only | Systemic only without skin | Skin and systemic | P |
|---|---|---|---|---|---|
| N | 25 | 5 | 5 | 15 | |
| Time from Dx to transplant, median (range), d | 182 (101, 335) | 189 (101, 271) | 156 (119, 169) | 189 (122, 335) | .091 |
| Time from Trt1 to transplant, median (range), d | 131 (72, 280) | 98 (72, 189) | 117 (108, 149) | 173 (72, 280) | .30 |
| Stage at transplant: CR | 25 (100) | 5 (100) | 5 (100) | 15 (100) | — |
| Type of transplant | |||||
| Autologous | 5 (20) | 3 (60) | 0 (0) | 2 (13) | .075 |
| Allogeneic | 20 (80) | 2 (40) | 5 (100) | 13 (87) | |
| Cell source | |||||
| Peripheral blood | 23 (92) | 5 (100) | 5 (100) | 13 (87) | — |
| Cord blood | 2 (8) | 0 (0) | 0 (0) | 2 (13) | |
| Donor type | |||||
| Autologous | 5 (20) | 3 (60) | 0 (0) | 2 (13) | — |
| MRD | 7 (28) | 0 (0) | 2 (40) | 5 (33) | |
| MURD | 5 (20) | 1 (20) | 1 (20) | 3 (20) | |
| MMURD | 6 (24) | 1 (20) | 2 (40) | 3 (20) | |
| Haploidentical | 1 (4) | 0 (0) | 0 (0) | 1 (7) | |
| Umbilical cord blood | 1 (4) | 0 (0) | 0 (0) | 1 (7) | |
| Conditioning regimen | |||||
| BEAM ± alemtuzumab | 4 (16) | 2 (40) | 0 (0) | 2 (13) | — |
| Bu/Cy | 1 (4) | 0 (0) | 0 (0) | 1 (7) | |
| Bu/Flu ± other | 5 (20) | 2 (40) | 0 (0) | 3 (20) | |
| CBV | 1 (4) | 0 (0) | 0 (0) | 1 (7) | |
| Clofarabine ± other | 3 (12) | 0 (0) | 1 (20) | 2 (13) | |
| Cy/Flu ± other | 4 (16) | 0 (0) | 1 (20) | 3 (20) | |
| Cy ± other | 3 (12) | 1 (20) | 1 (20) | 1 (7) | |
| Flu/Mel ± other | 4 (16) | 0 (0) | 2 (40) | 2 (13) | |
| GVHD prophylaxis | |||||
| Cyclo/MMF | 3 (12) | 0 (0) | 1 (20) | 2 (13) | — |
| TCD | 3 (12) | 0 (0) | 1 (20) | 2 (13) | |
| TBI/ATG | 1 (3) | 0 (0) | 0 (0) | 1 (7) | |
| Tac ± MMF or + Siro/MMF | 3 (10) | 0 (0) | 0 (0) | 3 (20) | |
| Tac/MTX | 4 (16) | 1 (20) | 2 (40) | 1 (7) | |
| Tac/MTX/Bort | 1 (3) | 0 (0) | 0 (0) | 1 (7) | |
| Tac/MTX/Siro | 4 (16) | 1 (20) | 1 (20) | 2 (13) | |
| Autologous/none | 5 (20) | 3 (60) | 0 (0) | 2 (13) | |
| Unknown | 1 (3) | 0 (0) | 0 (0) | 1 (7) |
| Characteristic | Total | Skin only | Systemic only without skin | Skin and systemic | P |
|---|---|---|---|---|---|
| N | 25 | 5 | 5 | 15 | |
| Time from Dx to transplant, median (range), d | 182 (101, 335) | 189 (101, 271) | 156 (119, 169) | 189 (122, 335) | .091 |
| Time from Trt1 to transplant, median (range), d | 131 (72, 280) | 98 (72, 189) | 117 (108, 149) | 173 (72, 280) | .30 |
| Stage at transplant: CR | 25 (100) | 5 (100) | 5 (100) | 15 (100) | — |
| Type of transplant | |||||
| Autologous | 5 (20) | 3 (60) | 0 (0) | 2 (13) | .075 |
| Allogeneic | 20 (80) | 2 (40) | 5 (100) | 13 (87) | |
| Cell source | |||||
| Peripheral blood | 23 (92) | 5 (100) | 5 (100) | 13 (87) | — |
| Cord blood | 2 (8) | 0 (0) | 0 (0) | 2 (13) | |
| Donor type | |||||
| Autologous | 5 (20) | 3 (60) | 0 (0) | 2 (13) | — |
| MRD | 7 (28) | 0 (0) | 2 (40) | 5 (33) | |
| MURD | 5 (20) | 1 (20) | 1 (20) | 3 (20) | |
| MMURD | 6 (24) | 1 (20) | 2 (40) | 3 (20) | |
| Haploidentical | 1 (4) | 0 (0) | 0 (0) | 1 (7) | |
| Umbilical cord blood | 1 (4) | 0 (0) | 0 (0) | 1 (7) | |
| Conditioning regimen | |||||
| BEAM ± alemtuzumab | 4 (16) | 2 (40) | 0 (0) | 2 (13) | — |
| Bu/Cy | 1 (4) | 0 (0) | 0 (0) | 1 (7) | |
| Bu/Flu ± other | 5 (20) | 2 (40) | 0 (0) | 3 (20) | |
| CBV | 1 (4) | 0 (0) | 0 (0) | 1 (7) | |
| Clofarabine ± other | 3 (12) | 0 (0) | 1 (20) | 2 (13) | |
| Cy/Flu ± other | 4 (16) | 0 (0) | 1 (20) | 3 (20) | |
| Cy ± other | 3 (12) | 1 (20) | 1 (20) | 1 (7) | |
| Flu/Mel ± other | 4 (16) | 0 (0) | 2 (40) | 2 (13) | |
| GVHD prophylaxis | |||||
| Cyclo/MMF | 3 (12) | 0 (0) | 1 (20) | 2 (13) | — |
| TCD | 3 (12) | 0 (0) | 1 (20) | 2 (13) | |
| TBI/ATG | 1 (3) | 0 (0) | 0 (0) | 1 (7) | |
| Tac ± MMF or + Siro/MMF | 3 (10) | 0 (0) | 0 (0) | 3 (20) | |
| Tac/MTX | 4 (16) | 1 (20) | 2 (40) | 1 (7) | |
| Tac/MTX/Bort | 1 (3) | 0 (0) | 0 (0) | 1 (7) | |
| Tac/MTX/Siro | 4 (16) | 1 (20) | 1 (20) | 2 (13) | |
| Autologous/none | 5 (20) | 3 (60) | 0 (0) | 2 (13) | |
| Unknown | 1 (3) | 0 (0) | 0 (0) | 1 (7) |
BEAM, carmustine, etoposide, cytarabine, melphalan; Bu/Cy, busulfan, cyclophosphamide; Bu/Flu, busulfan, fludarabine; CBV, cyclophosphamide, carmustine, etoposide; CR, complete remission; Cy, cyclophosphamide; Cy/Flu, cyclophosphamide, fludarabine; Cyclo/MMF, cyclosporine, mycophenolate mofetil; Dx, diagnosis; Flu/Mel, fludarabine, melphalan; MMURD, mismatched unrelated donor; MRD, matched, related donor; MURD, matched, unrelated donor; Siro, sirolimus; Siro/MMF, sirolimus, mycophenolate mofetil; Tac ± MMF, tacrolimus ± mycophenolate mofetil; Tac/MTX, tacrolimus, methotrexate; Tac/MTX/Bort, tacrolimus, methotrexate, bortezomib; Tac/MTX/Siro, tacrolimus, methotrexate, sirolimus; TBI/ATG, total body irradiation, antithymocyte globulin; TCD, T-cell depletion; Trt1, first-line therapy.
Outcome summary
| Variable | Total | Skin only | Systemic only without skin | Skin and systemic | Incomplete staging | P* |
|---|---|---|---|---|---|---|
| N, total | 54 | 11 | 10 | 27 | 6 | |
| OS 1 y % (95% CI) | 70 (55-81) | 90 (47-98) | 60 (25-83) | 64 (42-79) | 100 | .17 |
| 2 y % (95% CI) | 49 (34-62) | 80 (41-95) | 30 (7-58) | 43 (23-61) | 50 (1-91) | |
| Initiated treatment, N | 44 | 10 | 8 | 25 | 1 | |
| PFS 1 y % (95% CI) | 55 (39-68) | 80 (41-95) | 38 (9-67) | 52 (31-69) | 0 | .36 |
| 2 y % (95% CI) | 34 (21-48) | 60 (25-83) | 13 (1-42) | 32 (15-50) | 0 | |
| Transplanted, N† | 25 | 5 | 5 | 15 | 0 | |
| OS 1 y % (95% CI) | 80 (58-91) | 80 (20-97) | 80 (20-97) | 80 (50-93) | — | .69 |
| 2 y % (95% CI) | 60 (38-76) | 80 (20-97) | 40 (5-75) | 60 (32-80) | — | |
| N, allogeneic transplant | 20 | 2 | 5 | 13 | ||
| Cumulative incidence 1 y aGVHD grade 2-4, % (95% CI) | 42 (20-63) | 100 | 60 (7-91) | 31 (9-57) | — | .36 |
| aGVHD grade 2-4 | 9 (45) | 2 (100) | 3 (60) | 4 (31) | — | — |
| Skin | 2 | 1 | 0 | 1 | ||
| Skin and liver | 1 | 1 | 0 | 0 | ||
| Skin and lower GI | 1 | 0 | 0 | 1 | ||
| Skin and upper GI | 2 | 0 | 1 | 1 | ||
| Upper GI | 2 | 0 | 2 | 0 | ||
| Lower and upper GI | 1 | 0 | 0 | 1 | ||
| Cumulative incidence 1 y cGVHD, % (95% CI) | 20 (6-40) | 0 | 20 (4-63) | 23 (5-49) | — | .79 |
| cGVHD | 4 (20) | 0 (0) | 1 (20) | 3 (23) | — | — |
| Muscle and myositis | 1 | 0 | 0 | 1 | ||
| Liver and lower GI | 1 | 0 | 0 | 1 | ||
| Lung | 1 | 0 | 0 | 1 | ||
| Kidneys | 1 | 0 | 1 | 0 |
| Variable | Total | Skin only | Systemic only without skin | Skin and systemic | Incomplete staging | P* |
|---|---|---|---|---|---|---|
| N, total | 54 | 11 | 10 | 27 | 6 | |
| OS 1 y % (95% CI) | 70 (55-81) | 90 (47-98) | 60 (25-83) | 64 (42-79) | 100 | .17 |
| 2 y % (95% CI) | 49 (34-62) | 80 (41-95) | 30 (7-58) | 43 (23-61) | 50 (1-91) | |
| Initiated treatment, N | 44 | 10 | 8 | 25 | 1 | |
| PFS 1 y % (95% CI) | 55 (39-68) | 80 (41-95) | 38 (9-67) | 52 (31-69) | 0 | .36 |
| 2 y % (95% CI) | 34 (21-48) | 60 (25-83) | 13 (1-42) | 32 (15-50) | 0 | |
| Transplanted, N† | 25 | 5 | 5 | 15 | 0 | |
| OS 1 y % (95% CI) | 80 (58-91) | 80 (20-97) | 80 (20-97) | 80 (50-93) | — | .69 |
| 2 y % (95% CI) | 60 (38-76) | 80 (20-97) | 40 (5-75) | 60 (32-80) | — | |
| N, allogeneic transplant | 20 | 2 | 5 | 13 | ||
| Cumulative incidence 1 y aGVHD grade 2-4, % (95% CI) | 42 (20-63) | 100 | 60 (7-91) | 31 (9-57) | — | .36 |
| aGVHD grade 2-4 | 9 (45) | 2 (100) | 3 (60) | 4 (31) | — | — |
| Skin | 2 | 1 | 0 | 1 | ||
| Skin and liver | 1 | 1 | 0 | 0 | ||
| Skin and lower GI | 1 | 0 | 0 | 1 | ||
| Skin and upper GI | 2 | 0 | 1 | 1 | ||
| Upper GI | 2 | 0 | 2 | 0 | ||
| Lower and upper GI | 1 | 0 | 0 | 1 | ||
| Cumulative incidence 1 y cGVHD, % (95% CI) | 20 (6-40) | 0 | 20 (4-63) | 23 (5-49) | — | .79 |
| cGVHD | 4 (20) | 0 (0) | 1 (20) | 3 (23) | — | — |
| Muscle and myositis | 1 | 0 | 0 | 1 | ||
| Liver and lower GI | 1 | 0 | 0 | 1 | ||
| Lung | 1 | 0 | 0 | 1 | ||
| Kidneys | 1 | 0 | 1 | 0 |
aGVHD, acute GVHD; cGVHD, chronic GVHD; GI, gastrointestinal.
Test excludes patients in the Incomplete staging column.
Excludes 1 patient with no date of transplant or follow-up information.
Survival outcomes
A total of 54 patients had sufficient follow-up information to calculate OS, and 34 patients had died at the time of analysis. The median follow-up time for the 20 patients remaining alive was 2.6 years (range, 0.02-15.7 years). Dividing the cohort into approximately equal groups, there was no difference in survival between patients diagnosed in 2011 or earlier compared with those diagnosed later (P = .41). Table 4 lists the outcome for each group: OS from diagnosis (n = 54), PFS for those initiating therapy (n = 44), and OS for those undergoing transplant (n = 25). The median OS of the entire cohort was 24 months or 2.0 years (95% CI, 1.4-3.5) (Figure 1A). For those undergoing transplant, median OS from time of transplant was 6.6 years (95% CI, 1.64-NE [where NE signifies that the upper bound was not estimable]) (supplemental Figure 2).
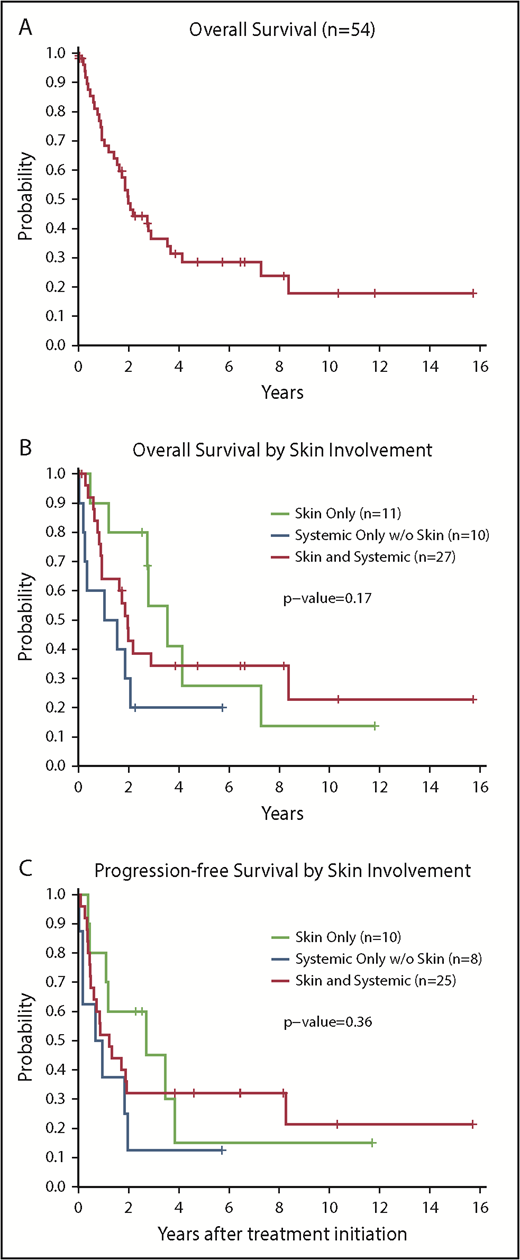
Survival outcomes according to organ involvement at diagnosis. (A) OS of the entire cohort (N = 54). (B) OS according to organ involvement at diagnosis (n = 48). (C) PFS (n = 43) according to organ involvement at diagnosis (test excludes patients with incomplete staging). OS and PFS were compared between groups by using a log-rank test. All P values reported are 2-sided and considered significant if <.05.
Survival outcomes according to organ involvement at diagnosis. (A) OS of the entire cohort (N = 54). (B) OS according to organ involvement at diagnosis (n = 48). (C) PFS (n = 43) according to organ involvement at diagnosis (test excludes patients with incomplete staging). OS and PFS were compared between groups by using a log-rank test. All P values reported are 2-sided and considered significant if <.05.
In this analysis, we identified no significant differences in OS (P = .17), PFS (P = .36), or survival after transplant (P = .69) in patients based on site of involvement at diagnosis (Figure 1B-C). Despite the sex bias in disease incidence (80% male in this cohort), there were no differences in survival based on sex (P = .76) (Figure 2A). However, patients aged <60 years had significantly longer survival than those aged ≥60 years (2-year OS, 70% vs 41%; P = .036) (Figure 2B). There was no difference in OS between those aged <60 years vs those aged ≥60 years among the 25 patients who received a transplant (P = .46) or for the subset of 20 patients who received an allogeneic transplant (P = .32).
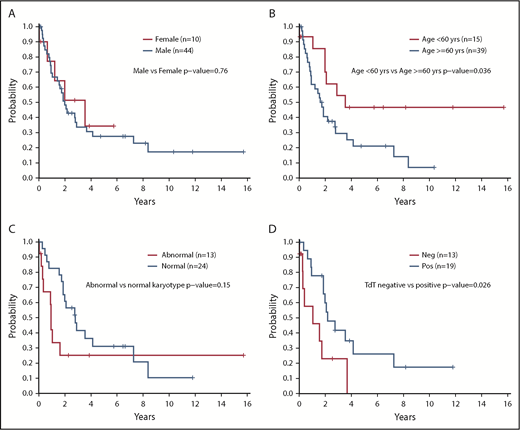
Survival outcomes according to patient characteristics and molecular data. (A) OS according to sex (N = 54). (B) OS according to age ≥60 years or <60 years (N = 54). (C) OS according to normal karyotype vs abnormal karyotype (n = 37); excludes 17 unknowns. (D) OS according to presence of TdT staining at diagnosis positive (Pos) vs negative (Neg) (n = 32); excludes 22 unknowns. OS was compared between groups by using a log-rank test. All P values reported are 2-sided and considered significant if <.05.
Survival outcomes according to patient characteristics and molecular data. (A) OS according to sex (N = 54). (B) OS according to age ≥60 years or <60 years (N = 54). (C) OS according to normal karyotype vs abnormal karyotype (n = 37); excludes 17 unknowns. (D) OS according to presence of TdT staining at diagnosis positive (Pos) vs negative (Neg) (n = 32); excludes 22 unknowns. OS was compared between groups by using a log-rank test. All P values reported are 2-sided and considered significant if <.05.
Having a normal karyotype was associated with a trend toward increased survival compared with any abnormal karyotype (2-year OS, 61% for normal vs 25% for abnormal; P = .15) (Figure 2C). Positive staining for terminal deoxynucleotidyltransferase (TdT) according to flow cytometry or immunohistochemistry was associated with improved survival vs a negative test result (2-year OS, 60% vs 23%; P = .026) (Figure 2D). Treatment with a lymphoid-type regimen (including all intensities) in first line showed a trend toward improved PFS compared with myeloid regimens (2-year PFS, 40% vs 11%; P = .075) (Figure 3A). Finally, the intensity of the treatment regimen (regardless of lymphoid or myeloid type) was also associated with survival, with intensive regimens having significantly increased PFS compared with nonintensive therapies (2-year PFS, 45% vs 11%; P = .034) (Figure 3B). Moderately intensive therapies had neither an advantage over nonintensive therapies nor a significant detriment compared with intensive therapies.
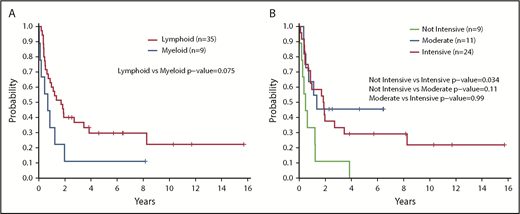
Survival outcomes according to treatment type and intensity. (A) PFS according to type of first-line therapy (lymphoid vs myeloid) (n = 44). (B) PFS according to intensity of first-line treatment (n = 44). PFS was compared between groups by using a log-rank test. All P values reported are 2-sided and considered significant if <.05.
Survival outcomes according to treatment type and intensity. (A) PFS according to type of first-line therapy (lymphoid vs myeloid) (n = 44). (B) PFS according to intensity of first-line treatment (n = 44). PFS was compared between groups by using a log-rank test. All P values reported are 2-sided and considered significant if <.05.
Responses to selected nonintensive treatments
Eight patients received treatment with single-agent pralatrexate in the course of therapy, including 3 as initial therapy and 5 at relapse. This was among the more common regimens used in this cohort, yet pralatrexate use was only previously reported in BPDCN in a single case report.24 The majority received pralatrexate 15 mg/m2 weekly (3 of 4 weeks), and 1 patient received 30 mg/m2 weekly (6 of 7 weeks). Two of the 3 patients treated with pralatrexate as first-line therapy achieved a complete remission and the third achieved a partial remission. The overall response rate to pralatrexate was 75% (6 of 8), with 50% (4 of 8) achieving complete remission and 25% (2 of 8) achieving partial remission. During treatment with pralatrexate, 63% (5 of 8) experienced adverse events (AEs) grade 2 or higher. Stomatitis and thrombocytopenia were observed in 38% (n = 3) and 25% (n = 2) of the patients, respectively. Other grade 2 AEs were nausea (n = 2), anemia (n = 1), neutropenia (n = 1), epistaxis (n = 1), and fatigue (n = 1), which are typical of pralatrexate therapy.25 Toxicities such as stomatitis were able to be mitigated by pralatrexate dose reductions or decreasing dose frequency.
One patient received treatment with the mutant isoform-selective IDH2 inhibitor enasidenib. This 79-year-old man had BPDCN involving skin and bone marrow at diagnosis, and he had comorbidities that precluded him receiving intensive chemotherapy. A targeted DNA-sequencing panel performed on bone marrow containing 91% BPDCN blasts revealed a DNA mutation encoding IDH2 R140W at 64.6% variant allele frequency. He received enasidenib at 100 mg daily; after 2 months of treatment, the patient’s bone marrow blasts were reduced to 32%, and the IDH2 mutant variant allele frequency had decreased to 23.6%. He experienced clinically stable disease with no significant drug-associated toxicities for ∼8 months before having disease progression in skin and bone marrow.
Discussion
By combining data between 3 major US cancer hospitals, we were able to increase our cohort size to one of the largest contemporary samples reported to date for this rare malignancy. The development of targeted agents specifically for BPDCN, such as tagraxofusp (SL-401), has galvanized the community of researchers and clinicians studying this disease.26 A number of additional agents directed against CD123,27 as well as other novel therapies, are in development for BPDCN. A “contemporary” historical data set such as this one has significant utility because it can serve to assist in evaluating emerging results from disease-specific trials of novel agents. However, the heterogeneity of the patient population in this observational study, particularly with regard to treatments received, limits it as a direct comparator cohort. Despite these caveats, because these data are from patients treated recently at the same centers now performing targeted therapy trials in BPDCN, we believe they address at least some of the biases inherent in other analyses.
Our data confirm that BPDCN is an aggressive malignancy associated with poor outcomes. The median OS of our cohort was at the higher end of what has been reported previously.28-30 This finding could be due to recent improvements in supportive care and diagnostic techniques, including improved recognition of BPDCN, compared with older data sets, although we observed no difference in survival between patients diagnosed in the early vs late portions of the cohort. It could also have resulted, in part, from a higher proportion of patients receiving an HSCT in this cohort compared with other published series. In any case, we believe our data represent an important reference for comparison with BPDCN trial cohorts because those studies are typically performed at the same or similar academic centers with access to transplantation.
An intriguing finding from our study was that BPDCN with “skin-only” involvement was not associated with better outcomes compared with systemic disease, although there was a trend toward higher median survival. A previous analysis reported that patients with skin-only BPDCN had improved survival compared with patients with other presentations.31 A definitive conclusion on outcomes in this subset may be limited by sample size or could be confounded by the longer time from diagnosis to treatment we observed in the skin-only patients (Table 2). In addition, we found that patients with skin-only disease were more likely to receive an autologous as opposed to an allogeneic HSCT. In theory, if BPDCN preleukemic or leukemia-initiating cells are present in morphologically “uninvolved” bone marrow, autologous stem cell products may transfer disease even in patients with apparent skin-only disease. However, despite this possibility, we observed patients with prolonged disease-free survival after autologous transplantation (supplemental Figure 2), consistent with previous reports.16 Thus, additional research is needed to understand disease ontogeny to determine how different sites of presentation should or should not influence treatment decisions, including transplantation.
TdT positivity in BPDCN cells was associated with a better OS compared with TdT negativity, consistent with a previous observation from a study of CD56+ hematologic malignancies before publication of the 2008 definition of BPDCN.32 Another study showed no survival association with TdT but a correlation with CD303 and high proliferative index33 ; we did not have sufficient cases with data for these markers to statistically evaluate them in our cohort. It is interesting that, despite the expression of typical lymphoid markers such as CD4 and TdT, the somatic mutations in BPDCN are more typical for myeloid malignancies.34-36 However, our findings, similar to those reported by others,29,37,38 suggest that patients who receive lymphoid-type chemotherapy regimens may experience better outcomes. Our cohort was not powered to determine if lymphoid marker expression correlated with response to lymphoid-type therapy. TdT could be evaluated as a prospective prognostic and/or predictive biomarker in future studies by using novel agents and/or conventional chemotherapy. Given these features, and other data such as RNA-sequencing, which suggested that the malignant cells were most similar to plasmacytoid dendritic cells or a close precursor,3,39 BPDCN biology may be distinct from either lymphoid or myeloid malignancies. This scenario provides another argument for studying BPDCN as a unique disease, both in the laboratory and in clinical trials.
In addition to novel agents targeting CD123, such as tagraxofusp (also including anti-CD123 monoclonal antibody-drug conjugates, CD3 × CD123 bispecific antibodies, and CD123 CAR T cells),27 there are other targets with promise in BPDCN. We previously found that BPDCN is particularly dependent on the antiapoptotic protein BCL-2 and highly sensitive to the oral BCL-2 inhibitor venetoclax,40 which has led to a phase 1 BPDCN-specific trial (NCT03485547). Additional agents with activity and tolerability in the BPDCN population may also be useful. Here, we found that the antifolate pralatrexate, approved by the US Food and Drug Administration for relapsed/refractory T-cell lymphomas,25,41-44 had efficacy in first-line and relapsed BPDCN and was relatively well tolerated. Together, our data indicate additional strategies that could be tested prospectively in BPDCN, including combinations of active conventional or targeted therapies with tagraxofusp. Optimal regimens may be based on age and comorbidity, such as using intensive vs nonintensive conventional chemotherapy, or by using biomarkers to select novel agents being developed in other malignancies, supported by the response to enasidenib in an IDH2-mutant BPDCN reported here.
Overall, our study provides a contemporaneously treated cohort of patients with BPDCN to help serve as the benchmark and comparator for the next generation of drugs in development for this disease. We found that age >60 years, abnormal karyotype, and negative staining for TdT were associated with poorer OS and, therefore, might be markers for testing risk-adapted therapy in the future. In agreement with previous reports, intensive therapies and stem cell transplantation were associated with improved PFS. However, in this unselected patient cohort, only 55% of patients received intensive therapy and 42% underwent HSCT, likely due to age or comorbidity. With the approval of tagraxofusp, the first drug with a specific indication for BPDCN, and the development of other agents with activity but improved tolerability compared with conventional chemotherapy, we remain hopeful for improved outcomes in patients with this disease.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
J.T. is supported by the Conquer Cancer Foundation of the American Society of Clinical Oncology, the American Association for Cancer Research, the American Society of Hematology, the Robert Wood Johnson Foundation, and the National Cancer Institute of the National Institutes of Health (1K08CA230319-01). A.A.L. is supported by the National Cancer Institute of the National Institutes of Health (5R37CA225191-01) and was an American Society of Hematology Scholar.
Authorship
Contribution: J.T., M.H., V.A.U., E.G., N.M.-S., A.M.B., A.L., S.B.L., A.D., A.T.F., R.M.S., M.S.T., R.K.R., S.M.H., and A.A.L. collected patient clinical data; D.S.N. and K.E.S. performed statistical analysis; and J.T. and A.A.L. designed the study and wrote the manuscript.
Conflict-of-interest disclosure: N.M.-S. has received research funding from Roche/Genentech, Celgene, Verastem, AstraZeneca, and Bristol-Myers Squibb and is a consultant for Kyowa-Hakka-Kirin. R.K.R. has received research funding from Incyte, Stemline Therapeutics, and Constellation and is a consultant for Incyte, Blueprint, Celgene, Agios, Jazz, BeyondSpring, Apexx, and Partner Therapeutics. S.M.H. has received research funding from Forty-Seven, Aileron, ADCT Therapeutics, Celgene, Infinity/Verastem, Millennium/Takeda, Seattle Genetics, and Trillium, and is a consultant for Kyowa-Hakka-Kirin, Millennium/Takeda, Infinity/Verastem, Seattle Genetics, Celgene, Miragen, Innate Pharma, Portola, Affimed, Kura, Mundipharma, and Astex. A.A.L. has received research funding from AbbVie and Stemline Therapeutics and is a consultant for N-of-One. The remaining authors declare no competing financial interests.
The current affiliation for E.G. is University of Southern California Keck School of Medicine Norris Comprehensive Cancer Center, Los Angeles, CA.
The current affiliation for N.M.-S. is Washington University School of Medicine, St. Louis, MO.
Correspondence: Andrew A. Lane, Dana-Farber Cancer Institute, 450 Brookline Ave, Mayer 413, Boston, MA 02215; e-mail: andrew_lane@dfci.harvard.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal