

Background: Genetic variants resulting in quantitative defects in the production of α-spectrin are associated with autosomal recessive hereditary spherocytosis (HS). Because erythroid cells produce α-spectrin in a 3-4:1 ratio in relation to β-spectrin, one defective allele is typically well-tolerated and a single null allele combined with a second partially defective allele may display a sub-clinical phenotype. Protein expression can be reduced by nonsense, frameshift, missense or splicing variants but reported large deletion events are extraordinarily rare (n = 1). Splicing variants include the low expression alleles αLELY - Low Expression LYon (SPTA1:c.6531-12C>T, p.?) and αLEPRA-Low Expression PRAgue (SPTA1:c.4339-99C>T, p.?) which reduce protein expression by 50% and 16%, respectively. Here, we present a family study with a whole gene SPTA1 deletion that characterizes the patient phenotype when a full gene mutation is co-inherited with the αLEPRA and αLELY low expression alleles.
Methods: The 6 month-old male proband had a history of congenital Coombs negative hemolytic anemia requiring transfusion from 3 months of age and in whom a red cell membrane disorder was clinically suspected. Pretransfusion CBC and peripheral blood smear results were unknown. Transfused CBC indices were as follows: RBC 4 x 1012/L, HGB 11 g/dL, MCV 76 fL, MCH 27 pg, MCHC 36 g/dL and a reticulocyte count of 1.8%. The proband's mother, of Cuban descent, and father, of European descent, had no history of anemia or HS. Targeted high throughput sequencing of genes related to hereditary hemolytic anemia was performed for the detection of single nucleotide variants (SNVs) and small insertion/deletion variants (indels). Relative quantitative real time PCR (RT-PCR) was utilized to confirm large structural variants.
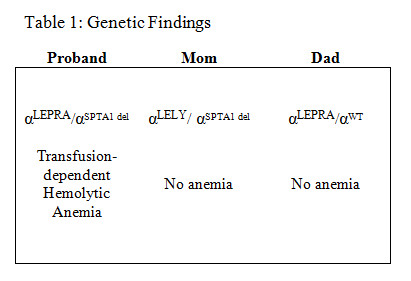
Results: Sequencing identified a large heterozygous deletion that spanned the entire SPTA1 gene (NC_000001.11:g.(?_158656337)_(158581024_?) in the proband and his mother. In addition, the proband had inherited hemizygous αLEPRA from his father. The mother had hemizgous αLELY. The father showed heterozygous αLEPRA and no other SPTA1 variants (Table 1).
Conclusion: To our knowledge, only Iolascon et al in 2011 has reported a SPTA1 gene large deletion (SpectrinExeter, exon 11-exon 12 deletion, loss of 83 amino acids) which resulted in a structurally abnormal protein and ellipto-poikilocytosis phenotype when in trans to αLELY. Our current case is the first reported SPTA1 full gene deletion and our family study allows comparison of the effects of coinheritance of the low expression alleles αLEPRA and αLELY. When in combination with complete absence of the SPTA1 gene in trans configuration, αLELY manifests in subclinical effects and αLEPRA causes a recessive HS phenotype, which resulted in severe anemia. This contrast further demonstrates that, unlike αLEPRA, αLELY does not decrease the α-spectrin concentration sufficiently to contribute to the baseline phenotype regardless of the severity of the second allele mutation for quantitative disorders such as HS.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal