

Key Points
MKL1-deficient neutrophils show severe functional defects due to defective actin polymerization and downregulation of actin-related proteins.
MKL1 deficiency does not alter primary fibroblast morphology, F-actin content, or migratory behavior.

Abstract
Megakaryoblastic leukemia 1 (MKL1) promotes the regulation of essential cell processes, including actin cytoskeletal dynamics, by coactivating serum response factor. Recently, the first human with MKL1 deficiency, leading to a novel primary immunodeficiency, was identified. We report a second family with 2 siblings with a homozygous frameshift mutation in MKL1. The index case died as an infant from progressive and severe pneumonia caused by Pseudomonas aeruginosa and poor wound healing. The younger sibling was preemptively transplanted shortly after birth. The immunodeficiency was marked by a pronounced actin polymerization defect and a strongly reduced motility and chemotactic response by MKL1-deficient neutrophils. In addition to the lack of MKL1, subsequent proteomic and transcriptomic analyses of patient neutrophils revealed actin and several actin-related proteins to be downregulated, confirming a role for MKL1 as a transcriptional coregulator. Degranulation was enhanced upon suboptimal neutrophil activation, whereas production of reactive oxygen species was normal. Neutrophil adhesion was intact but without proper spreading. The latter could explain the observed failure in firm adherence and transendothelial migration under flow conditions. No apparent defect in phagocytosis or bacterial killing was found. Also, monocyte-derived macrophages showed intact phagocytosis, and lymphocyte counts and proliferative capacity were normal. Nonhematopoietic primary fibroblasts demonstrated defective differentiation into myofibroblasts but normal migration and F-actin content, most likely as a result of compensatory mechanisms of MKL2, which is not expressed in neutrophils. Our findings extend current insight into the severe immune dysfunction in MKL1 deficiency, with cytoskeletal dysfunction and defective extravasation of neutrophils as the most prominent features.
Introduction
Megakaryoblastic leukemia 1 (MKL1) belongs to the myocardin-related transcription factor family, which also includes MKL2 and myocardin. These proteins function as coactivators of serum response factor (SRF), a highly conserved nuclear transcription factor.1-3
MKL1 is expressed in a wide range of embryonic and adult tissues.4 Within the cell, MKL1 is found in the cytoplasm in complex with globular actin (G-actin). Actin polymerization liberates MKL1 from G-actin, thereby allowing translocation of MKL1 to the nucleus, where it can activate SRF-dependent transcription. SRF regulates the expression of numerous genes that are involved in essential cell processes, including actin cytoskeletal dynamics, cell survival, and proliferation.5,6 Dysregulation of MKL1 in humans is implicated in the pathogenesis of cancer and cardiovascular diseases,5 and MKL1-knockout mice exhibit partial embryonic lethality, reduced platelet counts, and abnormal mammary gland function.7-9
In 2015, Record et al described the first human case with MKL1 deficiency caused by a homozygous nonsense mutation.10 The loss of MKL1 resulted in a primary immunodeficiency (PID) characterized by increased susceptibility to severe bacterial infections. Reduced F-actin levels were observed in neutrophils, peripheral blood mononuclear cell–derived dendritic cells, and an HL-60 myeloid leukemia MKL1-knockdown cell line.
In this article, we report on the severe phenotype, with a more in-depth and extensive analysis of primary neutrophil and fibroblast (dys)function in 2 novel MKL1-deficient infants. MKL1 deficiency in hematopoietic cells results in a pronounced actin polymerization defect and impaired chemotactic response of neutrophils; however, in nonhematopoietic cells (fibroblasts), the migratory behavior and F-actin content seemed to be normal, possibly as a result of the compensatory role of closely related MKL2, which is not expressed in neutrophils. Mass spectrometry of patient neutrophils and subsequent transcriptomics data further support a more complex role for MKL1 in actin metabolism beyond its binding to G-actin and involvement in actin polymerization.
Materials and methods
Isolation of human primary cells
Heparinized venous blood was drawn from healthy controls and from the MKL1-deficient patients after informed consent had been obtained. Blood from the index patient was collected on 3 occasions. Cord blood from the sibling was collected after caesarean delivery, and venous blood was collected on 1 occasion. Subsequently, neutrophils were isolated as described previously.11 The yield of the isolation was >90%. Purity was evaluated based on cytospins, which was >90% neutrophils; the remaining cells were eosinophils and lymphocytes. The cells were resuspended in HEPES medium12 for further functional testing. Although we cannot completely exclude preactivation during isolation, no increase in the neutrophil activation surface markers CD11b, CD66b, or CD63 was observed. Furthermore, no neutrophil activation was seen at baseline in functional experiments.
All experiments involving human blood samples were conducted in accordance with the Declaration of Helsinki. The study was approved by the local ethical committee of Sanquin Blood Supply, Amsterdam, The Netherlands and the Arnhem-Nijmegen Ethical Committee (nr.2010/104).
Additional methods
Statistical analysis and additional methods can be found in the supplemental Methods (available on the Blood Web site).
Results
Patient history and mutation analysis
The index case (male) was born as the second child to consanguineous parents from the Middle East at 36 weeks of gestation by emergency cesarean delivery because of partial placental abruption. During pregnancy, ultrasound showed polyhydramnios, macrosomia, and bilateral hydronephrosis. The patient had a mild asphyxia and recovered completely. At the age of 3.5 weeks he was admitted for respiratory insufficiency caused by pulmonary infection. Despite high-frequency oscillation ventilation and nitric oxide inhalation, he developed severe respiratory and circulatory failure. Veno-arterial extracorporeal membrane oxygenation (ECMO) was started at day 3. Initial investigations revealed leukocytosis (38.5 × 109/L) with normal erythrocyte and platelet counts, and a plasma C-reactive protein level of 91 mg/L that increased to 341 mg/L. Tracheal cultures were positive for Staphylococcus aureus and Pseudomonas aeruginosa and were treated with amoxicillin-clavulanic acid and ceftazidime. Four days later, polymerase chain reaction of a tracheal sample was positive for parainfluenza virus type 4, and cultures still showed P. aeruginosa, which had now become resistant to ceftazidime. Treatment was switched to meropenem, nebulized tobramycin, and ciprofloxacin. Corticosteroids were started to dampen the inflammatory response. Despite maximum supportive care, including IV immunoglobulin administration (in the presence of normal immunoglobulin G of 5.2 g/L), there was not a clinical or microbiological response. Although tracheal cultures remained positive for P. aeruginosa, all blood cultures remained negative. Laboratory investigations showed leukocytosis, initially with normal lymphocyte and neutrophil counts, which was rapidly followed by lymphopenia and thrombocytopenia. T-cell subsets were low (CD3, 0.6 × 109/L; CD4+, 0.45 × 109/L; CD8+, 0.14 × 109/L; ∼90% CD45RA+ phenotype). Attempts to wean from ECMO were unsuccessful. Bronchoscopic mucus evacuation and surfactant instillation did not result in any clinical improvement. Chest x-ray showed further deterioration, with extensive bullous emphysema. The umbilical cord had detached normally. On the other hand, an air leak from the venous cannula of the ECMO occurred as a result of poor wound healing. Because no improvement in the respiratory condition was seen after 4 weeks of ECMO, it was concluded that the lung disorder was irreversible, and continued ECMO was futile. After decannulation from ECMO, the patient deteriorated and died. A skin biopsy was done to perform fibroblastic culture; autopsy or post mortem lung biopsy was not allowed.
Because of clinical suspicion of neutrophil dysfunction, whole-exome sequencing was performed. A homozygous frameshift mutation [Chr22(GRCh37):g.40815086dup; NM_020831.4:c.1356dup (p.(Val453Argfs*16))] in MKL1 (Figure 1A) was identified. The absence of MKL1 protein in patient neutrophils was confirmed by western blotting (Figure 1B).
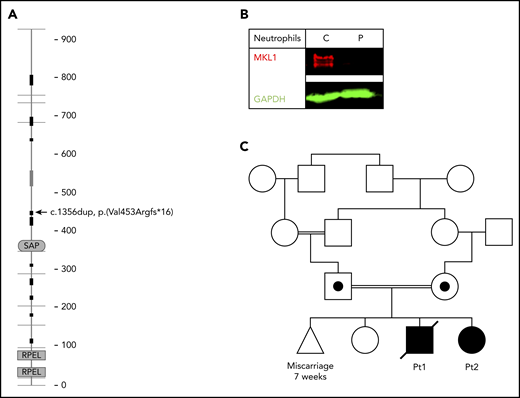
MKL1 mutation and family pedigree. (A) MKL1 protein structure; the location of the homozygous frameshift mutation leading to a premature stop codon is indicated (arrow) (adapted from SMART,68 with permission). (B) Absence of MKL1 in the patient’s neutrophils was confirmed by western blotting. GAPDH was used as loading control. (C) Pedigree of the family carrying the MKL1 mutation. Double lines indicate consanguinity; circle indicates female; square indicates male; triangle indicates miscarriage; diagonal through symbol indicates deceased; black symbols indicate clinically affected homozygous individual; black dot in symbol indicates clinically unaffected heterozygous individual. C, control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; P, patient.
MKL1 mutation and family pedigree. (A) MKL1 protein structure; the location of the homozygous frameshift mutation leading to a premature stop codon is indicated (arrow) (adapted from SMART,68 with permission). (B) Absence of MKL1 in the patient’s neutrophils was confirmed by western blotting. GAPDH was used as loading control. (C) Pedigree of the family carrying the MKL1 mutation. Double lines indicate consanguinity; circle indicates female; square indicates male; triangle indicates miscarriage; diagonal through symbol indicates deceased; black symbols indicate clinically affected homozygous individual; black dot in symbol indicates clinically unaffected heterozygous individual. C, control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; P, patient.
Analysis confirmed that both parents were heterozygous for this variant (for pedigree, see Figure 1C). One year later, the mother gave birth to another child (female, patient 2), who also was diagnosed with MKL1 deficiency. Prenatal diagnosis had been refused, and the pregnancy was uneventful. At birth, cord blood showed a mild thrombocytopenia (49 × 109/L), which was not observed in blood tests taken by subsequent venous punctures. Neutrophil and erythrocyte counts were normal, and thrombocyte counts remained stable at around 200 × 109/L, until this second affected sibling underwent a successful hematopoietic stem cell transplantation at the age of 9 weeks. At 8 months posttransplant, she had 100% chimerism and showed an adequate hematological and immunological recovery.
MKL1-deficient neutrophils have reduced actin levels and are defective in actin polymerization
Because human MKL1 deficiency has been described to result in a dysregulated actin cytoskeleton in neutrophils,10 we wanted to confirm these results. MKL1-deficient neutrophils indeed demonstrated a pronounced actin polymerization defect upon stimulation, as shown by flow cytometry and confocal microscopy analysis. Although control neutrophils displayed accumulation of F-actin (stained by phalloidin) at the leading edge upon stimulation with formyl-methionyl-leucyl phenylalanine (fMLF), MKL1-deficient neutrophils did not show detectable upregulation of F-actin. Basal F-actin levels were also found to be reduced compared with control neutrophils, and the levels of F-actin barely increased upon stimulation with C5a or fMLF, as assessed by flow cytometry with neutrophils from both patients and shown for the index case (Figure 2A-B). Expression levels of C5AR1 (receptor for C5a) and FPR1 and FPR2 (receptors for fMLF) were normal, as demonstrated by quantitative RNA analysis and mass spectrometry, confirming that our observations are not the result of a lack of receptor expression (supplemental Figure 1). Cytospins of the patient neutrophils showed segmented nuclei, indicative of mature neutrophils (Figure 2C). In addition to F-actin, total actin levels were reduced, as observed by western blotting (Figure 2D-E). Subsequent immuno-electron microscopy with immunogold labeling of actin confirmed a decrease in cortical and pseudopodial actin, and demonstrated that the patient’s pseudopodia were shorter and blunter in shape compared with those of control neutrophils (Figure 2F-G).
![MKL1-deficient neutrophils are defective in actin polymerization and have reduced actin levels. Actin polymerization was examined in suspension after stimulation with fMLF (10 nM) or C5a (10 nM) (mean ± range, n = 2 of the index patient) (A) and by confocal analysis with staining for F-actin (magenta = F-actin [phalloidin], cyan = DNA [Hoechst]) upon a 10-minute stimulation with fMLF (B). Images were acquired with a Leica TCS SP8 confocal microscope. Scale bars, 10 µm. (C) Cytospins of isolated neutrophils from control and index patient samples (original magnification ×400; May-Grünwald-Giemsa stain). (D) Reduced actin levels in MKL1-deficient neutrophils were detected using western blotting with anti-actin. GAPDH was used as loading control (n = 2 of the index case). (E) Quantification of actin levels in panel D. (F) Representative immuno-electron microscopy images stained for anti-actin (black dots). The pseudopodia were more bluntly shaped compared with control neutrophils (lower panels). Images were acquired with a Tecnai 12 G2 transmission electron microscope. Scale bars, 1 µm. (G) Quantification of gold-labeled particles in immuno-electron microscopy images stained for anti-actin (n = 1 of the index patient and control; 4 images were analyzed per donor). C, control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; P, patient.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/24/10.1182_blood.2019002633/3/m_bloodbld2019002633f2.png?Expires=1767733174&Signature=WVg6Qg9QdLePXyRpiCVdJM7khfAtabxFL~dpd4Z9Jbw-Wua6mHvfhQXNG5X1WqZmftAY8H2s25Gr7~8Ci79~ON9e6Ub3OsYDTg1GZVG1VjyHrXl-wdEUsQVN64PDBoGxEvW0k2RN1d6xHGACOxvMMMDSlXF1JEaI5cODusLpaoZoAPsQ1Y~YwK2tk9J7K80YlNaNsSZRKA5uAJpxGBrQzJnKWTl1EkktmG-SZOcvhRcVK5814oLuoEYL2XY6KY25yN7Pgy9QVqLRdNt1pL2Mu43CBMG47NjKQ9LtuQC5E3KkAK7B79~ovxWxiXcxMuqMRnx1erp5m1Xbrxgugf3Bzw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
MKL1-deficient neutrophils are defective in actin polymerization and have reduced actin levels. Actin polymerization was examined in suspension after stimulation with fMLF (10 nM) or C5a (10 nM) (mean ± range, n = 2 of the index patient) (A) and by confocal analysis with staining for F-actin (magenta = F-actin [phalloidin], cyan = DNA [Hoechst]) upon a 10-minute stimulation with fMLF (B). Images were acquired with a Leica TCS SP8 confocal microscope. Scale bars, 10 µm. (C) Cytospins of isolated neutrophils from control and index patient samples (original magnification ×400; May-Grünwald-Giemsa stain). (D) Reduced actin levels in MKL1-deficient neutrophils were detected using western blotting with anti-actin. GAPDH was used as loading control (n = 2 of the index case). (E) Quantification of actin levels in panel D. (F) Representative immuno-electron microscopy images stained for anti-actin (black dots). The pseudopodia were more bluntly shaped compared with control neutrophils (lower panels). Images were acquired with a Tecnai 12 G2 transmission electron microscope. Scale bars, 1 µm. (G) Quantification of gold-labeled particles in immuno-electron microscopy images stained for anti-actin (n = 1 of the index patient and control; 4 images were analyzed per donor). C, control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; P, patient.
MKL1-deficient neutrophils are defective in actin polymerization and have reduced actin levels. Actin polymerization was examined in suspension after stimulation with fMLF (10 nM) or C5a (10 nM) (mean ± range, n = 2 of the index patient) (A) and by confocal analysis with staining for F-actin (magenta = F-actin [phalloidin], cyan = DNA [Hoechst]) upon a 10-minute stimulation with fMLF (B). Images were acquired with a Leica TCS SP8 confocal microscope. Scale bars, 10 µm. (C) Cytospins of isolated neutrophils from control and index patient samples (original magnification ×400; May-Grünwald-Giemsa stain). (D) Reduced actin levels in MKL1-deficient neutrophils were detected using western blotting with anti-actin. GAPDH was used as loading control (n = 2 of the index case). (E) Quantification of actin levels in panel D. (F) Representative immuno-electron microscopy images stained for anti-actin (black dots). The pseudopodia were more bluntly shaped compared with control neutrophils (lower panels). Images were acquired with a Tecnai 12 G2 transmission electron microscope. Scale bars, 1 µm. (G) Quantification of gold-labeled particles in immuno-electron microscopy images stained for anti-actin (n = 1 of the index patient and control; 4 images were analyzed per donor). C, control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; P, patient.
To obtain additional supportive evidence for the findings in primary neutrophils, we assessed the consequence of loss of MKL1 in CD34+ hematopoietic stem cells that were differentiated toward the neutrophil lineage (for cytospins, see supplemental Figure 2A). Using an approach described previously,13 CRISPR-Cas9 editing of these cells resulted in a reduction in MKL1 levels of from 35% up to 74% compared with Cas9 control cells (supplemental Figure 2B-C; supplemental Figure 3 for the CRISPR-Cas9 model). Having a parallel culture for a knockout of a surface marker (ie, CD45), we have an estimation that ∼50% of the cells were complete knockout mixed with ~ 50% wild-type cells (supplemental Figure 4). Although not totally absent, as a result of the knockout of MKL1, we could confirm that total actin levels in the MKL1-knockout cultures were reduced, consistent with MKL1-deficient primary neutrophils (data not shown). When gated for the most mature neutrophil-like cells (ie, CD11b+CD16+ cells) in this mixed MKL1-knockout and wild-type culture, a decreased induction of F-actin upon stimulation with C5a was observed compared with control cells, as expected (supplemental Figure 2D).
Actin cytoskeletal-associated proteins and genes are downregulated in MKL1 deficiency
MKL1 functions as a coactivator of SRF, a highly conserved nuclear transcription factor.14 To further assess the consequence of MKL1 deficiency in neutrophils, high-resolution label-free quantitative mass spectrometry was performed on neutrophils from the index case (patient 1), the MKL1-deficient sibling (patient 2), and 3 healthy controls. Each experimental group consisted of 3 technical replicates.
Principal component analysis (PCA) showed 3 clusters: healthy control neutrophils, patient 1 neutrophils, and patient 2 neutrophils (Figure 3A). The difference in clustering between the 2 siblings might be explained by the fact that patient 1 was treated extensively at the time of sampling and was several months old, whereas patient 2 was a newborn who did not receive any treatment. Of the 2847 identified and quantified proteins with 3 valid values in ≥1 sample group, 40 proteins were significantly differentially expressed between patients and controls. Fourteen of these were significantly downregulated in both patients, including several actin-related proteins (ie, AIP1 [also known as WDR1], ARHGAP9, PFN1, and actin itself) (Figure 3B). Twenty-six proteins were upregulated in MKL1-deficient patients, which included several family members of the minichromosome maintenance proteins 2-7 (MCM2-7), a well-described hexamer complex involved in DNA replication.15,16
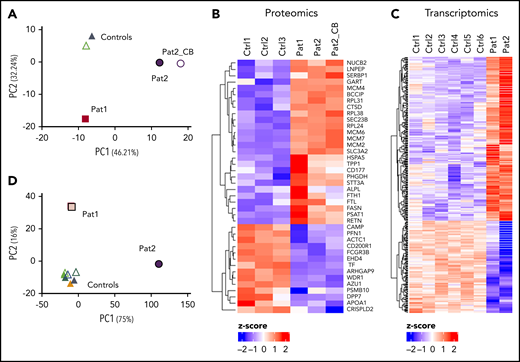
Proteomics and transcriptomic analysis showing differential protein and gene expression between patient and control neutrophils. PCA (A) and heat map (B) of proteome data. Heat map (C) and PCA (D) of transcriptome data. CB, neutrophils isolated from cord blood, Ctrl, control; Pat, patient.
Proteomics and transcriptomic analysis showing differential protein and gene expression between patient and control neutrophils. PCA (A) and heat map (B) of proteome data. Heat map (C) and PCA (D) of transcriptome data. CB, neutrophils isolated from cord blood, Ctrl, control; Pat, patient.
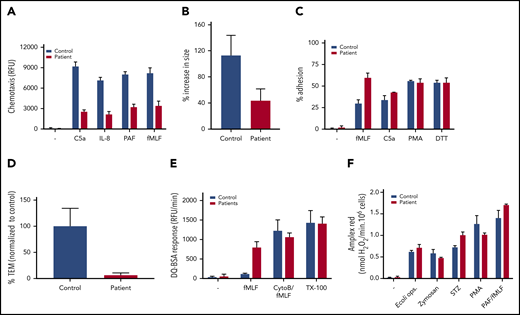
Neutrophil function tests. (A) Chemotaxis upon stimulation with C5a (10 nM), IL-8 (10 nM), PAF (100 nM), and fMLF (30 nM) measured using 3-µm pore–sized filters (mean + range, n = 2 of the index patient). (B) MKL1-deficient neutrophils show defective spreading behavior upon a 10-minute stimulation with fMLF (10 nM). Neutrophil spreading is quantified as the percentage of increase in size (mean + SEM, n = 1, 13-19 cells counted per condition). (C) Adhesion as percentage of total input upon stimulation with fMLF (1 µM), C5a (10 nM), PMA (100 ng/mL), or dithiothreitol (DTT, 1 mM) (mean + range, n = 2 of the index case). (D) Transendothelial migratory events were quantified and normalized to control neutrophils. Neutrophils from patient 2 were used (mean + standard error of the mean; n = 1, duplicate). (E) Protease release upon stimulation with fMLF (1 µM) or CytoB:fMLF (5 µg/mL:1 µM) or in a Triton X-100 lysate (TX-100) was determined by DQ Green BSA (mean + range, n = 3, both patients). (F) Extracellular reactive oxygen species production in response to opsonized E coli (0.25 × 109/mL), zymosan (1 mg/mL), serum-treated zymosan (STZ; 1 mg/mL), PMA (100 ng/mL), or PAF/fMLF (1 µM/1 µM) was measured using an Amplex Red assay (mean + range, n = 2, index patient).
Neutrophil function tests. (A) Chemotaxis upon stimulation with C5a (10 nM), IL-8 (10 nM), PAF (100 nM), and fMLF (30 nM) measured using 3-µm pore–sized filters (mean + range, n = 2 of the index patient). (B) MKL1-deficient neutrophils show defective spreading behavior upon a 10-minute stimulation with fMLF (10 nM). Neutrophil spreading is quantified as the percentage of increase in size (mean + SEM, n = 1, 13-19 cells counted per condition). (C) Adhesion as percentage of total input upon stimulation with fMLF (1 µM), C5a (10 nM), PMA (100 ng/mL), or dithiothreitol (DTT, 1 mM) (mean + range, n = 2 of the index case). (D) Transendothelial migratory events were quantified and normalized to control neutrophils. Neutrophils from patient 2 were used (mean + standard error of the mean; n = 1, duplicate). (E) Protease release upon stimulation with fMLF (1 µM) or CytoB:fMLF (5 µg/mL:1 µM) or in a Triton X-100 lysate (TX-100) was determined by DQ Green BSA (mean + range, n = 3, both patients). (F) Extracellular reactive oxygen species production in response to opsonized E coli (0.25 × 109/mL), zymosan (1 mg/mL), serum-treated zymosan (STZ; 1 mg/mL), PMA (100 ng/mL), or PAF/fMLF (1 µM/1 µM) was measured using an Amplex Red assay (mean + range, n = 2, index patient).
Although MKL1 was detected by western blot in control neutrophils (but not in patient neutrophils), mass spectrometry of neutrophils was unable to detect MKL1, in patient or in control neutrophils. Gene Ontology term enrichment analysis and nonimputed values of the significant proteins can be found in supplemental Table 1 and in supplemental Figures 5 and 6, respectively.
RNA sequencing was also performed to investigate the transcriptome of MKL1-deficient neutrophils (Figure 3C). A total of 14 740 genes was identified, of which 268 genes were significantly upregulated and 150 genes were significantly downregulated in patients compared with controls. PCA showed control neutrophils clustering together; again, the MKL1-deficient patients were found in separate clusters, possibly because of the substantial medical care that patient 1 was receiving (Figure 3D). The relative expression of MKL1 messenger RNA (mRNA) was decreased in patients, although it was not completely absent (data not shown).
There was a series of genes for which the transcriptional activity was significantly reduced or even absent in MKL1-deficient neutrophils compared with normal neutrophil transcriptome data. The transcription of genes related to adhesion processes, including CASS4 (regulates focal adhesion kinase and cell spreading),17 ALCAM (CD166),18 and EPHB1,19,20 and interferon signaling, including STAT1 and several IFIT genes, seemed to be dependent on MKL1 as a nonredundant transcriptional coactivator. Also in mice, knockdown of Srf in macrophages has been shown to inhibit the induction of several IFIT genes.21 A number of downregulated genes in the MKL1-deficient patients, including MYL9, have previously been described to be SRF target genes, confirming the role of MKL1 in transcriptional activity as an SRF coactivator.2,5,22-25
We also observed significantly upregulated transcript levels in MKL1-deficient neutrophils for the genes encoding neutrophil granule proteins (eg, PRTN3, MPO, BPI, and MMP8) (supplemental Table 2), although changes at the protein level in our proteomics data were not significant.
STRING interaction networks,26 a list of downregulated and upregulated gene transcripts, and Gene Ontology term enrichment analysis of the significantly altered genes can be found in supplemental Figure 7 and in supplemental Tables 2 and 3, respectively.
Together with the observed reduction in actin levels in these cells, the proteome and transcriptome data may well explain the loss of normal actin dynamics in MKL1-deficient neutrophils.
MKL1 deficiency results in impaired chemotaxis and neutrophil spreading
Because all higher organisms are totally dependent on a functional and well-regulated actin cytoskeletal rearrangement, we next wanted to assess the chemotactic response of MKL1-deficient neutrophils. We observed a strongly reduced chemotactic response through 3-µm pore– sized filters in response to C5a, interleukin-8 (IL-8), platelet-activating factor (PAF), and fMLF (Figure 4A). This observation was supported by TAXIScan imaging analysis; a clear chemotaxis defect is seen in response to C5a and fMLF (supplemental Video 1). Furthermore, confocal analysis showed the defective spreading behavior of neutrophils upon stimulation with fMLF on glass coverslips (Figure 4B). This corresponds with the electron microscopy data, which showed an altered pseudopodia phenotype in the patient (Figure 2F). Irrespective of the spreading defect, the adhesion capacity of MKL1-deficient neutrophils was not decreased under these static conditions (Figure 4C). We were unable to assess the adhesion capacity of the neutrophils of the index case under flow conditions, but we did observe a lack of leukocyte transendothelial migration under physiological flow conditions with neutrophils isolated from blood of his sibling (patient 2; Figure 4D). These MKL1-deficient cord blood neutrophils showed an identical F-actin defect (data not shown) and proteomic profile (Figure 3B) as neutrophils isolated from venous blood. Although MKL1-deficient neutrophils showed rolling over tumor necrosis factor α–stimulated endothelial cells, the cells were unable to firmly adhere, spread, or transmigrate through the endothelial monolayer under physiological flow conditions (Figure 4D; supplemental Video 2).
Antimicrobial functions of neutrophils in MKL1 deficiency
The index case suffered from a very early fatal Pseudomonas pneumonia while being treated with multiple broad-spectrum antibiotics to which the pathogen was sensitive. Hence, we performed a series of experiments to assess whether there was a dysfunction in 1 of the major antimicrobial mechanisms that neutrophils use to kill pathogens (ie, the process of microbial uptake, liberation of antimicrobial peptides and proteases from granules, and production of reactive oxygen species).27,28
In contrast to the case reported in 2015 by Record et al,10 we did not observe a defect in the ability of MKL1-deficient neutrophils to phagocytose Escherichia coli, S aureus, or serum-treated zymosan particles. Also no phagocytosis defect was observed in patient monocyte-derived macrophages (supplemental Figure 8A-B).
We found that protease release was enhanced upon suboptimal activation of MKL1-deficient neutrophils by fMLF (Figure 4E; supplemental Figure 2E for CD34+ MKL1-knockout cultures). No apparent difference was noted in the production of extracellular reactive oxygen species in response to particulate or soluble stimuli (Figure 4F).
Next, we tested the in vitro killing capacity of the neutrophils. Bacterial killing of E coli and S aureus, as well as killing of the yeast Candida albicans, was not defective when tested with the patient’s neutrophils (supplemental Figure 8C-D for the index patient).
In addition, we determined the patients’ T- and B-cell counts and subset distribution with ∼90% naive CD45+ T lymphocytes and no detectable CD27+ memory B cells and the intact in vitro proliferative capacity of these T and B cells toward CD3/CD28 and CpG/IL-2, respectively (data not shown).
Together, these results identified defective innate immunity as the primary defect in MKL1 deficiency caused by the impaired migratory capacity of innate immune cells.
Possible redundancy by MKL2 expression in MKL1-deficient fibroblasts
In addition to its role in hematopoietic cells, we speculated about the exact role of MKL1 activity in nonhematopoietic tissue cells. The effect of MKL1 deficiency on primary fibroblasts was assessed by various methods. First, Sanger sequencing confirmed the presence of the mutation in primary fibroblasts, and subsequent western blot analysis confirmed the complete absence of MKL1 protein in patient fibroblasts (Figure 5A). It has been previously reported that MKL1-knockout mice display defective myofibroblast differentiation.5,29,30
![Morphology and migration capacity of MKL1-deficient fibroblasts are not altered. (A) An absence of MKL1 in the index patient’s fibroblasts was observed by western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control. (B) Fibroblasts were left unstimulated or were stimulated with transforming growth factor-β (TGF-β; 10 ng/mL) for 48 hours, and αSMA was detected by western blotting. GAPDH was used as loading control. (C) Confocal analysis of fibroblasts by staining for F-actin (green = F-actin [phalloidin], cyan = DNA [Hoechst]). Images were acquired with a Leica TCS SP8 confocal microscope. Scale bar, 75 µm. (D) Actin levels in MKL1-deficient neutrophils were determined by western blotting with anti-actin. GAPDH was used as loading control. (E) Migration capacity of fibroblasts from the index case was assessed using a scratch assay (mean + range, n = 3). (F) Presence of MKL2 in control and patient fibroblasts was confirmed by western blotting, whereas MKL2 was not observed in neutrophils. αTubulin was used as loading control. Significance of fibroblast migration was tested using the Student t test. C, control; ns, not significant; P, patient.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/24/10.1182_blood.2019002633/3/m_bloodbld2019002633f5.png?Expires=1767733174&Signature=DjJEAwDKj0ovxWkDdYigR2WjeIU5cj7For6cEiY04IwAspuXJ9nOG1HFyV4GAJWXa8h96M1xL9RrdofF5sRzSn4GEhpztN1IrKurk7QBUD8BqViKUPAVGVcQLjkZVOvCoenS9T2qV~RNRkeL6jXOG6zxbkLuWlxlrErFdlYVpAaw2e6cTvv1~dDPUWKsbJ3vB4-6XLfZy89z3DIqv2njKsXTuRuIKu1jG1fq670qL1ia6ZyE62Tae1vi9UWkqn~10NrFo9iWgYKv-QJ-VCEzkmKOj4cxvLpxWl-SDPFFiQ2jR~0WZbSq3v~lsF03o6U4KJ-yFe7oRRiuZC~v~YD5ng__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Morphology and migration capacity of MKL1-deficient fibroblasts are not altered. (A) An absence of MKL1 in the index patient’s fibroblasts was observed by western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control. (B) Fibroblasts were left unstimulated or were stimulated with transforming growth factor-β (TGF-β; 10 ng/mL) for 48 hours, and αSMA was detected by western blotting. GAPDH was used as loading control. (C) Confocal analysis of fibroblasts by staining for F-actin (green = F-actin [phalloidin], cyan = DNA [Hoechst]). Images were acquired with a Leica TCS SP8 confocal microscope. Scale bar, 75 µm. (D) Actin levels in MKL1-deficient neutrophils were determined by western blotting with anti-actin. GAPDH was used as loading control. (E) Migration capacity of fibroblasts from the index case was assessed using a scratch assay (mean + range, n = 3). (F) Presence of MKL2 in control and patient fibroblasts was confirmed by western blotting, whereas MKL2 was not observed in neutrophils. αTubulin was used as loading control. Significance of fibroblast migration was tested using the Student t test. C, control; ns, not significant; P, patient.
Morphology and migration capacity of MKL1-deficient fibroblasts are not altered. (A) An absence of MKL1 in the index patient’s fibroblasts was observed by western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control. (B) Fibroblasts were left unstimulated or were stimulated with transforming growth factor-β (TGF-β; 10 ng/mL) for 48 hours, and αSMA was detected by western blotting. GAPDH was used as loading control. (C) Confocal analysis of fibroblasts by staining for F-actin (green = F-actin [phalloidin], cyan = DNA [Hoechst]). Images were acquired with a Leica TCS SP8 confocal microscope. Scale bar, 75 µm. (D) Actin levels in MKL1-deficient neutrophils were determined by western blotting with anti-actin. GAPDH was used as loading control. (E) Migration capacity of fibroblasts from the index case was assessed using a scratch assay (mean + range, n = 3). (F) Presence of MKL2 in control and patient fibroblasts was confirmed by western blotting, whereas MKL2 was not observed in neutrophils. αTubulin was used as loading control. Significance of fibroblast migration was tested using the Student t test. C, control; ns, not significant; P, patient.
Because the patient suffered from poor wound healing, we assessed fibroblast-to-myofibroblast differentiation, which is necessary for effective wound contraction and wound repair.31-33 Upon stimulation with transforming growth factor-β, MKL1-deficient fibroblasts showed no upregulation of α-smooth muscle actin (αSMA), a characteristic marker of myofibroblasts (Figure 5B). These data on impaired myofibroblast formation can be explained by the fact that the gene encoding αSMA, ACTA2, is an SRF-target gene.
Although the patient’s fibroblasts were lacking MKL1, we detected normal morphology, stress fibers of F-actin (examined on glass coverslips), total actin levels, and migratory behavior (as assessed by a scratch assay) of these nonhematopoietic primary cells (Figure 5C-E). These findings indicate that, in fibroblasts, there is either an insignificant role for MKL1 in cytoskeletal regulation and/or another transcriptional coactivator might render MKL1 redundant. We considered a role for MKL2 to compensate for the loss of MKL134,35 and investigated whether MKL2 is present in the patient’s fibroblasts. Indeed, MKL2 protein was detected by western blotting in cell lysates from control and patient fibroblast cultures. In contrast, mRNA quantification and western blot analysis revealed that no MKL2 is present in neutrophils to compensate for the loss of MKL1 in these cells (Figure 5F). Knockdown of MKL2 in patient primary fibroblasts with short hairpin RNA (shRNA) (Figure 6A-B) resulted in abnormal F-actin bundles in patient fibroblasts compared with fibroblasts expressing scrambled shRNA (Figure 6C). Furthermore, we observed that MKL2-knockdown fibroblasts (both control and patient) did not adhere and grow as well as control cells; however, no difference in morphology could be detected in MKL2-knockdown control fibroblasts by confocal imaging. These results indicate that loss of MKL1 or MKL2 in fibroblasts does not lead to profound alterations, whereas a loss of both proteins results in a dramatic loss of actin stress fibers. This might explain why the main clinical presentation of MKL1 deficiency seems to relate to a primary immunodeficiency of neutrophils, because MKL2 is not present in neutrophils.
![MKL2 knockdown in fibroblasts. (A) Knockdown of MKL2 in primary fibroblasts upon shRNA lentiviral transduction was assessed by western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control. (B) Quantification of MKL2 levels in panel A. (C) Confocal analysis of fibroblasts by staining for F-actin (green, F-actin [phalloidin]; cyan, DNA [Hoechst]). Images were acquired with a Leica TCS SP8 confocal microscope. Scale bars, 100 µm. SCR, scrambled.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/24/10.1182_blood.2019002633/3/m_bloodbld2019002633f6.png?Expires=1767733174&Signature=hSFc-akI1ObecfamiWTe58FxgsD-sRQhubmRfArEzqSi8wtt5wXFasnGWXkNo96HAxNvVTLHbWZ3stM~g1BybWBbCKEPcoB1oEnToAtNWBuacPn~0lZ9ukJnEIaucbS911qn8g-2P-BTv786iXx6N0t854rFDRUQ4KnLqz1oVWQPsSCx0YK6VLxm2eSzFWRCQQWfshVcT4z4-WF0miQa5GOnNYuLFu497h~Biiw8ksqeXzRHqt4vEMFzNf3QOR~lHlNr0Vpn~76mFpk~TNJExRcPVC0C4s5mvkLgqFIa2Mm6SowMWftiWP-mQ4W0cF3Ir3IPvME0H1qEiE9uFbrNIA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
MKL2 knockdown in fibroblasts. (A) Knockdown of MKL2 in primary fibroblasts upon shRNA lentiviral transduction was assessed by western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control. (B) Quantification of MKL2 levels in panel A. (C) Confocal analysis of fibroblasts by staining for F-actin (green, F-actin [phalloidin]; cyan, DNA [Hoechst]). Images were acquired with a Leica TCS SP8 confocal microscope. Scale bars, 100 µm. SCR, scrambled.
MKL2 knockdown in fibroblasts. (A) Knockdown of MKL2 in primary fibroblasts upon shRNA lentiviral transduction was assessed by western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control. (B) Quantification of MKL2 levels in panel A. (C) Confocal analysis of fibroblasts by staining for F-actin (green, F-actin [phalloidin]; cyan, DNA [Hoechst]). Images were acquired with a Leica TCS SP8 confocal microscope. Scale bars, 100 µm. SCR, scrambled.
Discussion
MKL1, also called myocardin-related transcription factor A, is an actin-binding protein that shuttles between the cytoplasm and nucleus in response to cell activation. In the nucleus, MKL1 mediates various cellular functions as a coactivator of SRF. MKL1 deficiency results in a PID with a phagocyte defect, as reported in a single case identified thus far.10 Upon identification of a second pedigree, we investigated the effect of MKL1 deficiency on primary hematopoietic and nonhematopoietic cell functions in more detail. In neutrophils, MKL1 deficiency indeed resulted in a pronounced actin polymerization defect, which explains the strongly reduced chemotactic response of these cells and is in agreement with the first report on human MKL1 deficiency.10
Our proteome and transcriptome analysis of patient neutrophils revealed that several actin-related and cytoskeletal-associated proteins are indeed downregulated in these cells, which is compatible with and supplementary to the observed reduction in actin levels initially noted in these MKL1-deficient neutrophils. For instance, AIP1, encoded by WDR1, promotes the disassembly of actin filaments through the enhancement of cofilin-dependent actin filament dynamics.36,37 WDR1 has been indicated to be an SRF-MTRF target gene.2 Recently, mutations in WDR1 have been shown to cause lazy leukocyte syndrome, an actin-related PID.38-41 These patients suffer from recurrent infections and neutropenia, and their neutrophils show increased F-actin levels (indicating abnormal actin polymerization) and impaired chemotaxis.
Together with guanine nucleotide exchange factors, GTPase-activating proteins regulate the activity of Rho family GTPases.42,43 The Rho GTPase activating protein 9 (encoded by ARHGAP9) was found to be reduced in the patient cells. This Rho GTPase-activating protein has been reported to be involved in regulating adhesion of hematopoietic cells to extracellular matrix.44-46 The actin-binding protein profilin, encoded by PFN1, is a nucleotide exchange factor for actin and an important regulator of actin cytoskeletal remodeling.47,48 Reduced expression may translate into reduced migration.49 Similar to our results in neutrophils, Joy et al showed that loss of MKL1 results in a downregulation of profilin expression in HEK-293 cells (human embryonic kidney epithelial cell line) and MDA-231 cells (human breast cancer cell line).50
Our transcriptomic analysis showed that a number of SRF-target genes were significantly downregulated (eg, MYL9, ZEB1, and REV3L), confirming that MKL1 acts as a coactivator of the transcription factor SRF.2,5,51 Compared with the first report on MKL1 deficiency, in which cytoskeletal-specific polymerase chain reaction arrays were used, we only found MYL9 to be differentially expressed in MKL1-deficient cells. However, Record et al used MKL1-deficient dHL-60 cells instead of primary neutrophils.10
We found increased expression of the MCM hexamer complex, which has been demonstrated in numerous cancers.15,16,52,53 The protein expression profiles of all MCM subunits are downregulated in neutrophils during myeloid differentiation of progenitors into mature neutrophils,54 which suggests that the levels in circulating neutrophils represent young neutrophils in both infants, even though their nuclei already had the multilobular morphology and were behaving functionally as fully differentiated neutrophils (Figure 2C). In the same way, we may explain the upregulated transcript levels of some of the granule components (eg, PRTN3, MPO, BPI, and MMP8). Normally, the expression levels of these gene transcripts are elevated in the neutrophil precursors and drop during differentiation into segmented neutrophils.54,55
MKL1-deficient neutrophils showed a defective chemotactic response, impaired spreading behavior, and impaired transendothelial migration, whereas their adhesion capacity under static conditions was not altered. The normal integrin expression (CD11a, CD11b, and CD18) in MKL1-deficient neutrophils, as determined by flow cytometry and proteome and transcriptome analysis (data not shown), is in agreement with the findings in mouse neutrophils that completely lack the transcription factor Srf.22 Binding of the extracellular domain of integrins to their ligand might be sufficient for neutrophils to initially adhere, whereas the subsequent conformational change of integrins and association with cytoplasmic integrin-actin linking proteins (eg, talin and vinculin, which are encoded by MKL1-SRF target genes5 ) are necessary for adhesion strengthening and cell spreading.22,56-58 The index case exhibited leukocytosis at presentation, a hallmark of leukocyte adhesion deficiencies (LADI and LADIII).59,60 However, the sibling had a normal neutrophil count, as did the patient described by Record et al10 ; therefore, the leukocytosis is best explained by severe infection. This supports the finding that MKL1 deficiency does not alter static adhesion capacity, suggesting that inside-out signaling of integrins might be still intact, whereas outside-in signaling is impaired.
The index case suffered from ongoing Pseudomonas pneumonia, despite treatment with antibiotics to which the infecting strain was sensitive. A normal in vitro phagocytosis and killing capacity was found in the patient neutrophils and monocyte-derived macrophages. The susceptibility to bacterial infection in MKL1 deficiency can be explained by the inability of such neutrophils to migrate toward the site of infection. Comparable to our previous study on ARPC1B deficiency with a similar defect in neutrophil chemotaxis and actin polymerization,61 we hypothesize that MKL1-deficient neutrophils become activated and release their toxic products prematurely when sticking to the endothelial cell wall close to the site of infections, while being unable to transmigrate. This is supported by the observation that in vitro protease release by MKL1-deficient neutrophils was enhanced upon suboptimal cell activation. During this frustrated extravasation, the neutrophils may cause collateral damage (manifesting as vascular damage or vasculitis), locally at the site where the infection cannot be cleared or in a more widespread manner. Because MKL1-deficient neutrophils are able to phagocytose and kill microbes properly in vitro, this may also explain why all blood cultures remained negative for microbial growth, even in the absence of antibiotics. In contrast to ARPC1B deficiency,61 basal levels of F-actin and total actin levels in patient neutrophils were substantially reduced, which may be explained by the role of MKL1 in SRF-dependent transcription of SRF target genes, including actin.5,22
The actin stress fibers and total actin levels were normal in MKL1-deficient primary fibroblasts. This is in contrast to the first report on MKL1 deficiency, in which a loss of cortical actin and stress fibers was observed in fibroblasts. However, lacking primary fibroblasts from the patient, the investigators used MKL1-targeting shRNA in normal primary fibroblasts, which could explain the differences observed.10
Although our primary MKL1-deficient fibroblasts demonstrated defective differentiation into myofibroblasts, their morphology and migratory behavior were normal, possibly as a result of MKL2 expression in nonhematopoietic cells, because knockdown of MKL2 by shRNA resulted in a dramatic loss of actin stress fibers in MKL1-deficient fibroblasts. MKL1 and MKL2 are located on different chromosomes but share similar structural motifs4,62 and have been demonstrated to play redundant roles.34,35 Nonetheless, these compensatory mechanisms of MKL2 are not absolute, as indicated by defective differentiation of mammary myoepithelial cells in MKL1-knockout mice, despite normal MKL2 expression,9 and the defective development of fibroblasts into myofibroblasts observed in our study.
A number of PIDs have been identified that are caused by mutations in genes involved in regulation of the actin cytoskeleton, including ARPC1B deficiency, Wiskott-Aldrich syndrome, WDR1 deficiency, DOCK2 and DOCK8 deficiency, and leukocyte adhesion deficiencies (LADI, LADIII).40,59-61,63-67 These mutations primarily affect hematopoietic cells, because a number of actin-regulatory proteins are exclusively or predominantly expressed in immune cells. Although most PID caused by cytoskeletal defects can be classified as (severe) combined immunodeficiencies, MKL1 deficiency seems to manifest primarily as a severe phagocyte disorder, because T and B cells of the patient had intact proliferative capacity. Also, no T- or B-cell defect was reported in the previously described MKL1-deficient patient.10
Collectively, our findings demonstrate a nonredundant role for MKL1 in human neutrophils, with cytoskeletal dysfunction and reduced F-actin levels as key features. In fibroblasts, MKL2 expression, which is missing in neutrophils, seems to compensate for the absence of MKL1. With recent developments in whole-exome sequencing, more actin-related PIDs are expected to be identified, which will provide more knowledge about the role of the actin cytoskeleton in the human immune system.
The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository (dataset identifier PXD014633). The RNA sequencing data have been deposited in the Gene Expression Omnibus (accession number GSE134644).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the parents for their collaboration. They are grateful to Dirk Roos and Maartje van den Biggelaar for critically reading the manuscript. They also thank Paul Tuijnenburg and Machiel Jansen for extensive lymphocyte tests, Arjan Hoogendijk for assistance with RNA analysis, and Tuula Rinne for supervision of genomic diagnostics.
This study was funded, in part, by the European Union’s Horizon 2020 Research and Innovation Program under Grant Agreement No.668303, Program on Prevention Outcomes Practices Grant PPOP-12-001, the Center of Immunodeficiencies Amsterdam Grant CIDA-2015, and the E-Rare ZonMW grant 90030376506.
Authorship
Contribution: E.G.G.S., A.T.J.T., and T.W.K. designed the experiments; E.G.G.S., A.T.J.T., I.v.d.B., M.v.H., C.E.M.A., S.T., P.J.J.H.V., H.J.M.P.V., and C.W.B. performed experiments and analyzed the data; S.S.V.H. analyzed clinical data; J.G. and M.d.B. performed DNA/RNA analysis; I.C.K. and F.P.J.v.A. performed mass spectrometry analysis and RNA analysis; W.K. and T.G. performed whole-exome sequencing; H.J. performed electron microscopy and analyzed the results; E.G.G.S., S.S.V.H., and T.W.K. wrote the manuscript; and all authors read, revised, and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Evelien G. G. Sprenkeler, Department of Blood Cell Research, Sanquin Research, Amsterdam University Medical Center, University of Amsterdam, Amsterdam 1066CX, The Netherlands; e-mail: e.sprenkeler@sanquin.nl.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal