

Key Points
This first prospective trial of PD-1 blockade for post–alloHCT relapse showed activity primarily in lymphoid malignancies.
Minimal activity was seen in myeloid malignancies, and GVHD and immune-related adverse events occurred.

Abstract
Programmed cell death-1 (PD-1)/programmed death ligand-1 blockade may potentially augment graft-vs-tumor effects following allogeneic hematopoietic cell transplantation (alloHCT), but retrospective studies of anti–PD-1 therapy reported substantial toxicity from graft-versus-host-disease (GVHD). Here, we report the results of a prospective clinical trial of PD-1 blockade for relapsed hematologic malignancies (HMs) after alloHCT (NCT01822509). The primary objective in this phase 1 multicenter, investigator-initiated study was to determine maximum tolerated dose and safety. Secondary objectives were to assess efficacy and immunologic activity. Patients with relapsed HMs following alloHCT were eligible. Nivolumab was administered every 2 weeks until progression or unacceptable toxicity, starting with a 1-mg/kg cohort, with planned deescalation based on toxicity to a 0.5-mg/kg cohort. Twenty-eight patients were treated (n = 19 myeloid, n = 9 lymphoid). Median age was 57 years (range 27-76), and median time from alloHCT to enrollment was 21 months (range 5.6-108.5). Two of 6 patients treated at 1 mg/kg experienced dose-limiting toxicity (DLT) from immune-related adverse events (irAEs). Twenty-two patients were treated at 0.5 mg/kg, and 4 DLTs occurred, including 2 irAEs and 2 with fatal GVHD. The overall response rate in efficacy-evaluable patients was 32% (8/25). With a median follow-up of 11 months, the 1-year progression-free survival and overall survival were 23% and 56%, respectively. In this first prospective clinical trial of an anti–PD-1 antibody for post–alloHCT relapse, GVHD and irAEs occurred, requiring dose deescalation, with only modest antitumor activity. Further studies of anti–PD-1 therapy post–alloHCT may require specific toxicity mitigation strategies. This trial was registered at www.clinicaltrials.gov as #NCT 01822509.
Introduction
Relapse of hematologic malignancies (HMs) is the most common cause of death following allogeneic hematopoietic cell transplant (alloHCT) in the modern era.1 Therapeutic options for relapse have limited efficacy.2 Across the broad population of HMs, 3-year post–relapse overall survival (OS) is ∼20%,1 and patients with relapsed acute myeloid leukemia (AML) fare particularly poorly, with a 1-year post–relapse OS of 20% despite subsequent therapy.3 Thus, novel therapeutic approaches for this population are urgently needed.
Disease relapse may be partly attributed to tumor cell evasion of donor T-cell immunity. This may occur through engagement of inhibitory immune checkpoints through tumor cell expression of the inhibitory ligands B7-1/B7-2 and programmed death ligand-1/2 (PD-L1/PD-L2), which engage cytotoxic T-lymphocyte–associated protein 4 (CTLA-4) and programmed cell death ligand 1 (PD-1) receptors, respectively, on donor T cells. This impairs antitumor immunity through inducing T-cell exhaustion, downregulating cytotoxic T-cell (CTL) activity, and creating tumor niches, eventually leading to donor T-cell apoptosis.4-7
We previously studied CTLA-4 blockade for patients with HMs that relapsed post–alloHCT. After an initial dose-finding study,8 we demonstrated in a phase 1 study of ipilimumab that this approach is feasible and active for post–alloHCT relapse.9 About two-thirds of patients treated at the maximum tolerated dose (MTD) experienced disease reduction, including patients with both myeloid and lymphoid malignancies, suggesting that the therapeutic effect was mediated through an augmented graft-versus-tumor (GVT) effect. Although the treatment was tolerable for most, immune-related adverse events (irAEs) or graft-versus-host disease (GVHD) occurred in about a third of patients.
Blockade of PD-1 in alloHCT mouse models demonstrated potent antileukemic effects, although also exacerbated GVHD.10-12 Additional studies suggested that host PD-L1 is dominant over PD-L2 in regulating GVHD lethality,13 and PD-L1 expression on donor T cells may drive GVHD lethality.14 These preclinical data suggest that PD1 blockade may have efficacy for patients with relapsed HM post–alloHCT, although may also increase the risk of GVHD. They also suggest that the risks and benefits of targeting PD-1 following alloHCT may be different from those of targeting CTLA-4.
Two recent retrospective cohort studies found that PD-1 blockade for relapsed Hodgkin lymphoma (HL) after alloHCT led to dramatic antitumor activity; however, substantial toxicities were also observed due to GVHD, which in some cases proved to be severe and treatment refractory.15,16 Given the strong preclinical data for PD-1 blockade post–alloHCT, we hypothesized that this approach could be efficacious for a broad population of HMs. Because of the safety concerns with GVHD, it seemed critical to conduct a prospective clinical trial with dose exploration to assess whether a safe regimen could be identified. Here, we report the results of a multicenter, phase 1 clinical trial of nivolumab for patients with relapsed HMs after alloHCT.
Patients and methods
Eligibility
Patients aged ≥18 years with progressive or persistent disease after alloHCT and a diagnosis of one of the following were eligible: chronic lymphocytic leukemia, non-Hodgkin lymphoma, HL, multiple myeloma, AML, acute lymphoblastic leukemia, myelodysplastic syndrome (MDS), myeloproliferative neoplasm, or chronic myeloid leukemia (CML). The definition of progressive or persistent disease was per standard criteria for each diagnosis.17-23 An Eastern Cooperative Oncology Group (ECOG) performance status score of ≤2 was required. Participants were required to have donor T-cell chimerism of ≥20% at study entry and were excluded if they had received donor lymphocyte infusion within 8 weeks prior to registration or had a history of grade 3 or 4 acute GVHD (aGVHD) or active, severe chronic GVHD (cGVHD). Additional exclusion criteria were impaired liver or kidney function or receipt of other anticancer therapy or investigational agents within 4 weeks prior to registration. Patients with autoimmune disease or patients who required systemic immunosuppressive medications within 4 weeks prior to registration were also excluded.
Trial design and assessment of safety and response
This was an open-label, investigator-initiated, Cancer Therapy Evaluation Program (CTEP)-sponsored, phase 1 study of nivolumab conducted at 8 sites across the United States (CTEP 9204, #NCT01822509). Nivolumab was administered as an IV infusion on day 1 of an every-2-week cycle, with the first 8 cycles considered induction followed thereafter by maintenance therapy every 2 weeks until disease progression or unacceptable toxicity. After treatment discontinuation, patients were monitored for up to 1 year to capture potential late toxicities.
The dose-finding portion of the study followed a univariate binomial design with the primary objective of determining the MTD of nivolumab in this patient population. The expansion portion of the trial was designed to enroll 15 additional patients to confirm safety and tolerability at the MTD. Secondary objectives included determining the rate of disease response, progression-free survival (PFS), OS, and assessing immunologic correlates.
The starting dose level of the dose-finding portion was 1 mg/kg with possible deescalation to 0.5 mg/kg or escalation to 3 mg/kg. The lower starting dose of 1 mg/kg (compared with standard nivolumab dosing at 3 mg/kg for other diseases) was chosen to balance potential safety concerns with respect to GVHD with the risk of decreased efficacy with even lower dosing in this population that had exhausted other therapeutic options. Dose-limiting toxicities (DLTs) were defined as: stage 4 skin aGVHD; ≥ stage 3 gut or liver aGVHD; grade 4 hematologic toxicity considered unrelated to underlying disease that did not respond to growth factor or transfusion support within 7 days; ≥ grade 3 nonneurologic irAEs that did not respond to corticosteroid treatment within 3 weeks; and other ≥ grade 3 nonhematologic toxicity that did not improve to ≤ grade 1 after dose delay.
A lengthy DLT observation period of 12 weeks was incorporated due to the potential for immune-mediated toxicity to take several weeks to manifest. To minimize accrual delays, 5 patients were enrolled to each dose level. If ≤1 DLT was observed among 5 evaluable patients, the dose would be escalated. If >1 DLT was observed, the MTD would be considered exceeded, and the dose would be deescalated. A preplanned early stopping rule was incorporated for a 15-patient expansion cohort such that if ≥2 DLTs occurred among the first 5 patients, or ≥4 DLTs occurred among all 15 patients, then enrollment would cease.
Adverse events were evaluated using Common Terminology Criteria for Adverse Events, version 4.0 criteria, and all patients who received study treatment were considered evaluable for toxicity. All patients were assessed for disease response using standard disease-specific criteria after the completion of 8, 16, 32, and 48 weeks on therapy, and at treatment discontinuation. The trial was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice guidelines. Institutional review board approval was obtained at each site, and all patients provided informed consent. All authors had access to primary clinical trial data and attest that the trial was conducted in accordance with the protocol.
Correlative studies
Flow cytometry was performed on fresh whole blood samples collected at various times before and after treatment. Cells were stained with a panel of directly conjugated monoclonal antibodies to identify T cells, B cells, natural killer (NK) cells, and dendritic cells (DCs), as well as functional subsets (supplemental Table 4, available on the Blood Web site). Labeled cells were acquired in a FACSCanto II flow cytometer (BD Biosciences) and analyzed using BD FACSDiva software (BD Biosciences) using standardized gating strategies24 (also see supplemental Figures 1 to 3). To calculate absolute cell counts, a complete blood count on the same day as flow analysis provided the percentage of total cells comprised of lymphocytes and a calculated absolute lymphocyte count. T-, B-, and NK-cell analysis is gated on lymphocytes, and absolute counts for cells defined as % by flow markers were calculated from the absolute lymphocyte count. DC analysis is gated on all CD45+ cells, and absolute number of DCs is based on the total white blood cell count. Donor T-cell chimerism was calculated from the proportion of allele-specific donor-derived amplicon in extracted DNA. Identification of genetic malignant clones was by the Rapid Heme Panel, a next-generation sequencing platform, as previously described.25 Histopathologic and cytogenetic analyses of bone marrow specimens along with percentages of blasts and CD3+CD8+ T cells were conducted per routine clinical testing in the pathology laboratory at the Brigham and Women’s Hospital by expert hematopathologists.
Statistical analysis
The data cutoff for the statistical analysis was March 26, 2019. Descriptive statistics was employed for reporting continuous variables and frequency and percentage of discrete variables. The Kaplan-Meier method was used to estimate the secondary endpoints of PFS (defined as time from first nivolumab treatment to time of progression or death) and OS (defined as time from first nivolumab treatment to time of death). Univariable Cox regression analysis was employed to assess the effect of achieving complete remission (CR)/partial remission on OS and PFS, treating time to response as a time-dependent variable. Time between the latest alloHCT and study initiation in association with time to onset of acute or chronic (a/c) GVHD was assessed using the likelihood ratio test in a Cox model. Fisher’s exact test or Wilcoxon rank sum test was used to test possible associations between baseline characteristics and irAEs or response. Wilcoxon signed rank test and exact Wilcoxon rank sum test were used for paired and group comparison of immunophenotype data, respectively. All P values were 2-sided, and the significance level was set to .05. Multiplicity was not considered. All analyses were performed using SAS 9.3 (SAS Institute Inc, Cary, NC), and R version 3.2.2 (the CRAN project, www.cran.r-project.org). Heat maps were generated using GENE-E (http://www.broadinstitute.org/ cancer/software/GENE-E). Hierarchical clustering in heat map recursively merged objects based on their pairwise distance using the Spearman’s correlation for distance measures.
Results
Patient characteristics
A total of 28 patients received treatment between October 2016 and May 2018. Their baseline characteristics are detailed in Table 1, with additional details on the AML patients in supplemental Table 1. The most frequent primary diagnoses were AML (n = 10), MDS (n = 7), and HL (n = 5). Six patients had received myeloablative (21.4%) and 22 received reduced intensity (78.4%) conditioning. Twenty-two patients received peripheral blood stem cells (78.6%), 5 patients received bone marrow (17.9%), and 1 patient received both (3.6%). The median number of prior treatment regimens was 2 (range, 1 to 9), with a median time between alloHCT and trial registration of 21 months (range, 5.6 to 108.5). Prior to study entry, 68% of patients (n = 19) had received at least 1 prior therapy for relapse following alloHCT. Twelve patients had prior acute (n = 9) or chronic (n = 4) GVHD (one had both). All patients had been off immune suppression for at least 4 weeks prior to starting on study.
Baseline patient characteristics
| Characteristic (N = 28) | N | % |
|---|---|---|
| Age, median (range) | 57 (27, 76) | |
| Patient sex | ||
| Male | 21 | 75 |
| Female | 7 | 25 |
| ECOG performance status | ||
| 0 | 10 | 35.7 |
| 1 | 15 | 53.6 |
| 2 | 3 | 10.7 |
| Primary disease | ||
| AML | 10 | 35.7 |
| MDS | 7 | 25 |
| HL | 5 | 17.9 |
| Non-Hodgkin lymphoma* | 3 | 10.7 |
| Leukemia, NOS | 1 | 3.6 |
| Chronic lymphocytic leukemia | 1 | 3.6 |
| Myeloproliferative disorder† | 1 | 3.6 |
| Disease status at transplant | ||
| 1st CR | 9 | 32.1 |
| 2nd CR | 1 | 3.6 |
| Partial remission | 9 | 32.1 |
| Untreated | 1 | 3.6 |
| Refractory (induction failure) | 5 | 17.9 |
| Relapsed | 3 | 10.7 |
| HLA typing (A, B, C, DRB1, DQB1) | ||
| 8/8 matched | 19 | 67.9 |
| 6/6 matched | 20 | 71.4 |
| Donor type | ||
| Unrelated donor | 13 | 46.4 |
| Matched related donor | 12 | 42.9 |
| Haploidentical donor | 3 | 10.7 |
| Recipient/donor sex | ||
| Male ← Male | 16 | 57.1 |
| Male ← Female | 5 | 17.9 |
| Female ← Male | 4 | 14.3 |
| Female ← Female | 3 | 10.7 |
| Conditioning intensity | ||
| Nonmyeloablative | 22 | 78.6 |
| Myeloablative | 6 | 21.4 |
| Source of progenitor cells | ||
| Peripheral blood stem cells | 22 | 78.6 |
| Bone marrow | 5 | 17.9 |
| Bone marrow and PBSC | 1 | 3.6 |
| Disease status at time of enrollment | ||
| Relapsed | 23 | 82.1 |
| Persistent disease | 5 | 17.9 |
| Prior treatment of disease post–alloHCT | ||
| Yes | 19 | 67.9 |
| No | 9 | 32.1 |
| Prior aGVHD | ||
| Yes (3 grade 1, 6 grade 2) | 9 | 32.1 |
| No | 19 | 67.9 |
| Prior cGVHD | ||
| Yes (3 limited, 1 extensive) | 4 | 14.3 |
| No | 24 | 85.7 |
| Any prior a/c GVHD | ||
| Yes | 12 | 42.9 |
| No | 16 | 57.1 |
| Number of prior treatment regimens (excluding alloHCT), median (range) | 2 (1, 9) | |
| Prior autologous stem cell transplantation, patients (%) | 5 (17.9) | |
| Months from relapse to enrollment, median (range) | 1.1 (0.33, 38.4) | |
| Months from alloHCT to enrollment, median (range) | 21 (5.6, 108.5) | |
| Characteristic (N = 28) | N | % |
|---|---|---|
| Age, median (range) | 57 (27, 76) | |
| Patient sex | ||
| Male | 21 | 75 |
| Female | 7 | 25 |
| ECOG performance status | ||
| 0 | 10 | 35.7 |
| 1 | 15 | 53.6 |
| 2 | 3 | 10.7 |
| Primary disease | ||
| AML | 10 | 35.7 |
| MDS | 7 | 25 |
| HL | 5 | 17.9 |
| Non-Hodgkin lymphoma* | 3 | 10.7 |
| Leukemia, NOS | 1 | 3.6 |
| Chronic lymphocytic leukemia | 1 | 3.6 |
| Myeloproliferative disorder† | 1 | 3.6 |
| Disease status at transplant | ||
| 1st CR | 9 | 32.1 |
| 2nd CR | 1 | 3.6 |
| Partial remission | 9 | 32.1 |
| Untreated | 1 | 3.6 |
| Refractory (induction failure) | 5 | 17.9 |
| Relapsed | 3 | 10.7 |
| HLA typing (A, B, C, DRB1, DQB1) | ||
| 8/8 matched | 19 | 67.9 |
| 6/6 matched | 20 | 71.4 |
| Donor type | ||
| Unrelated donor | 13 | 46.4 |
| Matched related donor | 12 | 42.9 |
| Haploidentical donor | 3 | 10.7 |
| Recipient/donor sex | ||
| Male ← Male | 16 | 57.1 |
| Male ← Female | 5 | 17.9 |
| Female ← Male | 4 | 14.3 |
| Female ← Female | 3 | 10.7 |
| Conditioning intensity | ||
| Nonmyeloablative | 22 | 78.6 |
| Myeloablative | 6 | 21.4 |
| Source of progenitor cells | ||
| Peripheral blood stem cells | 22 | 78.6 |
| Bone marrow | 5 | 17.9 |
| Bone marrow and PBSC | 1 | 3.6 |
| Disease status at time of enrollment | ||
| Relapsed | 23 | 82.1 |
| Persistent disease | 5 | 17.9 |
| Prior treatment of disease post–alloHCT | ||
| Yes | 19 | 67.9 |
| No | 9 | 32.1 |
| Prior aGVHD | ||
| Yes (3 grade 1, 6 grade 2) | 9 | 32.1 |
| No | 19 | 67.9 |
| Prior cGVHD | ||
| Yes (3 limited, 1 extensive) | 4 | 14.3 |
| No | 24 | 85.7 |
| Any prior a/c GVHD | ||
| Yes | 12 | 42.9 |
| No | 16 | 57.1 |
| Number of prior treatment regimens (excluding alloHCT), median (range) | 2 (1, 9) | |
| Prior autologous stem cell transplantation, patients (%) | 5 (17.9) | |
| Months from relapse to enrollment, median (range) | 1.1 (0.33, 38.4) | |
| Months from alloHCT to enrollment, median (range) | 21 (5.6, 108.5) | |
Non-Hodgkin lymphoma patients included diffuse large B-cell lymphoma (n = 2) and PMBCL (n = 1).
Myeloproliferative disorder patients included 1 patient with CMML.
NOS, not otherwise specified; PBSC, peripheral blood stem cell.
MTD assessment
Six patients received treatment with nivolumab at 1 mg/kg (including one who had progressive disease prior to 12 weeks and came off study, and thus was not evaluable for DLT assessment). Two of these patients experienced irAEs that met DLT criteria: 1 case of sepsis leading to fatal acute respiratory distress syndrome thought to be exacerbated by drug-related immune activation (based on autopsy results), and 1 case of newly diagnosed anti–phospholipid antibody syndrome complicated by a fatal thrombotic cerebral vascular accident (Table 2). The dose of nivolumab was thus deescalated to 0.5 mg/kg in a subsequent cohort of 8 patients (including 3 with early progressive disease who came off study and were not evaluable for DLT). No DLTs were observed, and the MTD was determined to be 0.5 mg/kg.
Summary of dose-limiting toxicities
| Dose (mg/kg) | Toxicity | Grade | Outcome |
|---|---|---|---|
| 1.0 | Sepsis with acute respiratory distress syndrome | 5 | Fatal |
| 1.0 | APLS with CVA | 5 | Fatal |
| 0.5 | GVHD (liver and gut) | 5 | Fatal |
| 0.5 | GVHD (liver and gut) | 5 | Fatal |
| 0.5 | Elevated bilirubin | 3 | Resolved after >4 wk with drug hold, steroids |
| 0.5 | Transaminitis | 3 | Resolved after >4 wk with drug hold, steroids |
| Dose (mg/kg) | Toxicity | Grade | Outcome |
|---|---|---|---|
| 1.0 | Sepsis with acute respiratory distress syndrome | 5 | Fatal |
| 1.0 | APLS with CVA | 5 | Fatal |
| 0.5 | GVHD (liver and gut) | 5 | Fatal |
| 0.5 | GVHD (liver and gut) | 5 | Fatal |
| 0.5 | Elevated bilirubin | 3 | Resolved after >4 wk with drug hold, steroids |
| 0.5 | Transaminitis | 3 | Resolved after >4 wk with drug hold, steroids |
APLS, antiphospholipid syndrome; CVA, cerebrovascular accident.
Assessment of safety
Fourteen patients were subsequently enrolled to the expansion cohort at 0.5 mg/kg. Accrual was terminated 1 patient earlier than planned due to the occurrence of 4 additional DLTs, which met the protocol-defined stopping rule. These DLTs included stage 3 liver (n = 1) and gut (n = 1) aGVHD, grade 3 elevated bilirubin (n = 1), and grade 3 transaminitis (n = 1), which did not recover to ≤ grade 1 within 4 weeks of holding the drug and immunosuppressive therapy (Table 2). Both patients with immune-mediated liver dysfunction had no histological evidence of GVHD and eventually improved, but both patients with aGVHD died due to GVHD complications.
Patients received a median of 3 cycles of nivolumab therapy (range, 1 to 29), and at the time of the data cut, all patients were off treatment. During trial treatment or following treatment discontinuation, 11 patients (39%) developed new or worsening a/c-GVHD, including 2 patients with aGVHD, 8 patients with cGVHD (2 of whom had cGVHD at baseline), and 1 patients with both. Of 9 patients with new onset of a/c GVHD, shorter time between alloHCT and study initiation was observed (median time 12.7 months for new onset of a/c GVHD vs 32.2 months for no new GVHD; P = .02).
Additional serious adverse events beyond the DLTs previously described included, at 1 mg/kg: grade 3 pneumonitis, grade 3 transaminitis, and grade 3 respiratory syncytial virus pneumonia (n = 1 each), and at 0.5 mg/kg: grade 4 lipase elevation, grade 3 rash, grade 3 orthostatic hypotension, grade 3 transaminitis, and grade 2 seizure (in a patient with a history of seizure disorder) (n = 1 each). The most commonly occurring treatment-emergent adverse events of any grade are reported in Table 3 and included fatigue (n = 12, 43%), transaminitis (n = 8, 29%), and maculopapular rash (n = 8, 29%). The only factor associated with the development of irAEs was ECOG PS. Of the 10 patients with irAEs, 9 had a baseline PS of 1 or higher (P = .048). Time from alloHCT to study enrollment was shorter for patients who developed irAEs compared with those without irAEs, although the difference is not statistically significant (median time from BMT to enrollment 11.2 months vs 31 months, respectively; P = .11). There was also no association between GVHD prophylactic regimen and the development of irAEs. Treatment-emergent hematologic toxicity not attributed to the underlying HM was relatively uncommon (Table 3). Ten patients (36%) required dose delays due to toxicity. Reasons for nivolumab discontinuation included progressive disease (n = 12), unacceptable toxicity (n = 11), physician decision (n = 3), and patient decision (n = 2).
Summary of all-grade nonhematologic adverse events occurring in >10% of patients and all treatment-emergent hematologic adverse events
| Toxicity | Grade 1/2, n (%) | Grade 3, n (%) | Grade 4, n (%) | Total, n (%) |
|---|---|---|---|---|
| Nonhematologic | ||||
| Fatigue | 10 (36) | 2 (7.1) | 0 | 12 (43) |
| Rash | 7 (25) | 1 (3.6) | 0 | 8 (29) |
| Transaminitis | 6 (21) | 2 (7.1) | 0 | 8 (29) |
| Fever | 4 (14) | 1 (3.6) | 0 | 5 (18) |
| Lipase elevation | 1 (3.6) | 2 (7.1) | 2 (7.1) | 5 (18) |
| Pneumonitis | 3 (11) | 1 (3.6) | 0 | 4 (14) |
| Abdominal pain | 3 (11) | 1 (3.6) | 0 | 4 (14) |
| Nausea | 3 (11) | 1 (3.6) | 0 | 4 (14) |
| Vomiting | 4 (14) | 0 | 0 | 4 (14) |
| Arthralgia | 2 (7.1) | 1 (3.6) | 0 | 3 (11) |
| Bilirubin increase | 0 | 3 (11) | 0 | 3 (11) |
| Confusion | 2 (7.1) | 1 (3.6) | 0 | 3 (11) |
| Diarrhea | 3 (11) | 0 | 0 | 3 (11) |
| Dry mouth | 3 (11) | 0 | 0 | 3 (11) |
| Headache | 3 (11) | 0 | 0 | 3 (11) |
| Hematologic | ||||
| Thrombocytopenia | 2 (7.1) | 0 | 4 (14) | 6 (21) |
| Neutropenia | 1 (3.6) | 1 (3.6) | 3 (11) | 5 (18) |
| Anemia | 1 (3.6) | 3 (11) | 0 | 4 (14) |
| Febrile neutropenia | 0 | 2 (7.1) | 1 (3.6) | 3 (11) |
| Toxicity | Grade 1/2, n (%) | Grade 3, n (%) | Grade 4, n (%) | Total, n (%) |
|---|---|---|---|---|
| Nonhematologic | ||||
| Fatigue | 10 (36) | 2 (7.1) | 0 | 12 (43) |
| Rash | 7 (25) | 1 (3.6) | 0 | 8 (29) |
| Transaminitis | 6 (21) | 2 (7.1) | 0 | 8 (29) |
| Fever | 4 (14) | 1 (3.6) | 0 | 5 (18) |
| Lipase elevation | 1 (3.6) | 2 (7.1) | 2 (7.1) | 5 (18) |
| Pneumonitis | 3 (11) | 1 (3.6) | 0 | 4 (14) |
| Abdominal pain | 3 (11) | 1 (3.6) | 0 | 4 (14) |
| Nausea | 3 (11) | 1 (3.6) | 0 | 4 (14) |
| Vomiting | 4 (14) | 0 | 0 | 4 (14) |
| Arthralgia | 2 (7.1) | 1 (3.6) | 0 | 3 (11) |
| Bilirubin increase | 0 | 3 (11) | 0 | 3 (11) |
| Confusion | 2 (7.1) | 1 (3.6) | 0 | 3 (11) |
| Diarrhea | 3 (11) | 0 | 0 | 3 (11) |
| Dry mouth | 3 (11) | 0 | 0 | 3 (11) |
| Headache | 3 (11) | 0 | 0 | 3 (11) |
| Hematologic | ||||
| Thrombocytopenia | 2 (7.1) | 0 | 4 (14) | 6 (21) |
| Neutropenia | 1 (3.6) | 1 (3.6) | 3 (11) | 5 (18) |
| Anemia | 1 (3.6) | 3 (11) | 0 | 4 (14) |
| Febrile neutropenia | 0 | 2 (7.1) | 1 (3.6) | 3 (11) |
Assessment of efficacy
Twenty-five patients were evaluable for response (5 treated at 1 mg/kg; 20 treated at 0.5 mg/kg). Three patients were considered unevaluable because they experienced early toxicity without disease progression requiring discontinuation prior to the first response evaluation. The overall response rate (ORR) for the efficacy evaluable population across both dose levels was 32% (8/25, baseline characteristics of responders in supplemental Table 3). Of the 5 evaluable patients in the 1-mg/kg dose group, 1 patient achieved CR (primary mediastinal large B-cell lymphoma [PMBCL]) and 2 patients achieved PR (MDS and chronic myelomonocytic leukemia [CMML]). Of the 20 evaluable patients in the 0.5-mg/kg dose group, 5 patients responded, including 3 PRs in patients with HL and 2 PRs in patients with MDS. The intent-to-treat ORR for the entire study cohort was 29% (8/28), including 4 of 9 (44%) with lymphoid malignancies and 4 of 19 (21%) in myeloid malignancies (P = .37). Responders had a shorter time from alloHCT to study entry than nonresponders (median 11.8 months vs 31 months, P = .05). The ORR for patients who received reduced intensity conditioning was 36% (8/22), whereas none of the 6 patients who received myeloablative conditioning responded (P = .14). Response was not significantly associated with the development of irAEs, as 50% (5/10) patients with irAEs responded, whereas 17% (3/18) of patients without irAEs did not (P = .09). None of the 6 patients with extramedullary relapse of myeloid disease (including 2 with isolated CNS relapse only) responded to nivolumab.
With a median follow-up time among survivors of 11 months (range, 3.6 to 29), the median PFS in all 28 patients was 3.7 months (95% confidence interval [CI], 1.6, 8), with a 1-year PFS of 23% (95% CI, 8%, 43%) (Figure 1). The median OS in all 28 patients was 21.4 months (95% CI, 4.2, not reached), with a 1-year OS in all patients of 56% (95% CI, 35%, 72%) (Figure 1). The survival of patients with lymphoid and myeloid malignancies was compared, and although there was a trend toward improved survival for patients with lymphoid disease, this was not statistically significant for OS (Figure 2A; P = .06) or PFS (Figure 2B; P = .08). The impact of achieving CR/PR on PFS and OS was assessed in a univariable Cox model treating time to response as a time-dependent variable. The hazard ratio for CR/PR in PFS was 1.31 (95% CI, 0.41, 4.26; P = .65); the hazard ratio for CR/PR in OS was 0.66 (95% CI, 0.14, 3.18; P = .6). Two out of 8 responders and 11 out of 20 nonresponders died.
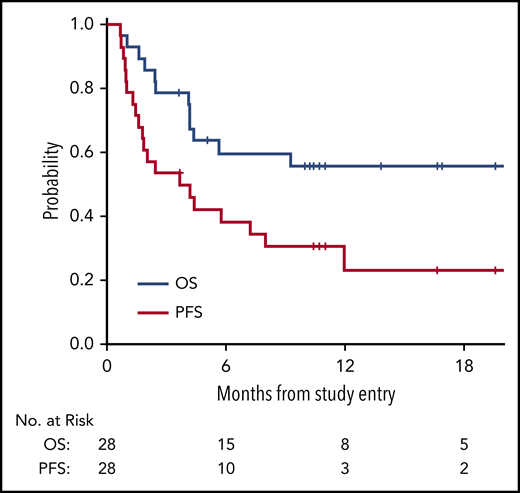
Kaplan-Meier survival curves assessed in all patients. OS curve (blue) and PFS curve (red) for all patients on trial (n = 28).
Kaplan-Meier survival curves assessed in all patients. OS curve (blue) and PFS curve (red) for all patients on trial (n = 28).
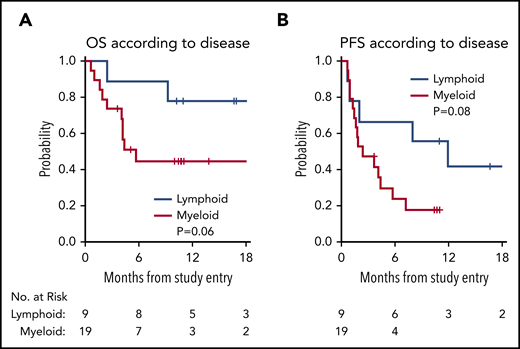
OS and PFS by lymphoid vs myeloid disease. (A) OS by lymphoid (blue) vs myeloid (red) disease. (B) PFS by lymphoid (blue) vs myeloid (red) disease.
OS and PFS by lymphoid vs myeloid disease. (A) OS by lymphoid (blue) vs myeloid (red) disease. (B) PFS by lymphoid (blue) vs myeloid (red) disease.
Assessment of immunologic correlates
Based on our prior work demonstrating that CTLA-4 blockade post–alloHCT could decrease activation of regulatory T cells (Treg) and expand conventional effector T-cell populations (Tcon) in the blood, immunophenotyping by flow cytometry was performed on serial blood samples freshly acquired from patients at the lead site. Analysis focused on 17 patients with a baseline sample and at least 1 subsequent sample (supplemental Table 2). Between baseline sampling and cycle 8, patients treated with nivolumab had an increase in circulating CD3+ T cells (P = .039), particularly CD4+ (P = .039) T cells and normal B cells (P = .05), whereas no increase was seen in NK cells (P = .38) nor in DCs (P = 1.0) (Figure 3A; supplemental Figure 4). A hierarchical clustering analysis was performed to evaluate whether the percentage of PD-1 positivity at baseline in T-cell subsets was associated with clinical response. Compared with patients with progressive disease, patients with CR/PR or stable disease (SD) had a higher baseline level of PD-1 expression on circulating Treg (P = .006) and CD4+ (P = .04) (Figure 3B). In addition, patients with CR/PR or SD had a lower ratio of Treg to total CD8+ T cells at cycle 4 (P = .014) (Figure 3C). In the patient with CMML who achieved PR (see patient 49 in supplemental Table 3 for more information), morphologic and cytogenetic response in the bone marrow was preceded by marrow expansion of CD3+CD8+ T cells (along with expansion of CD3+CD8+CD57+ CTLs in the peripheral blood, data not shown) and was accompanied by increased marrow donor cell chimerism (Figure 3D). Next-generation sequencing on sequential bone marrow samples from this patient revealed multiple genetic CMML clones and subclones with differential sensitivity to anti–PD-1 therapy, with a minimal residual RUNX1 (Q213*) subclone persisting as the sole marker of malignancy after nivolumab treatment (Figure 3E).
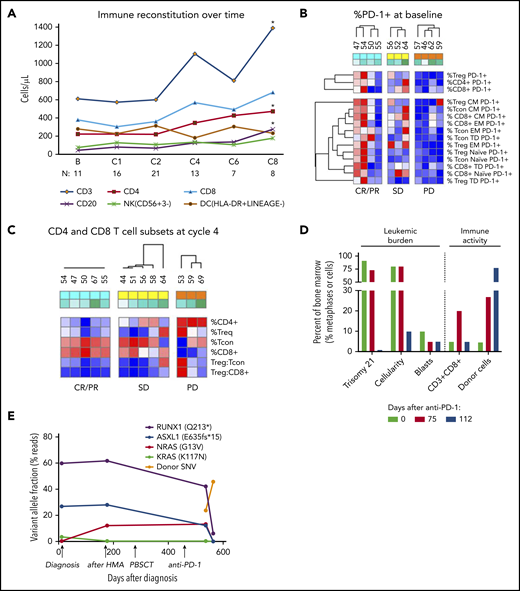
Assessment of immunologic correlative studies. (A) Time course of immune cell populations at baseline and on treatment through cycle 8 (C8), including total T cell (CD3+), CD4+, and CD8+ T cell, B cell (CD20+), NK cell (CD56+CD3−), and DC (HLA-DR+LINEAGE−). Each dot represents median value. *P ≤ .05 compared with the baseline value. (B) Baseline %PD-1 expression in T-cell subsets in patients who went on to achieve response (CR/PR; green), SD (yellow), or progressive disease (PD) (orange) as best response. Red color represents the maximum value, and the blue color represents the minimum value in each row. (C) CD4 and CD8 T-cell subsets at cycle 4 according to response status, including Treg and Tcon. Red color represents the maximum value, and the blue color represents the minimum value in each row. (D) % of morphologic and cytogenetic bone marrow involvement as well as immune activity from CD3+CD8+ T-cell and donor-cell chimerism in a responding patient with CMML at baseline (green), day +75 post nivolumab (red), and day +112 post nivolumab (blue). The % of trisomy 21 is 100% of 10 XY recipient metaphase cells that were evaluated by the clinical cytogenetics laboratory, while the % of CTLs was 20% of the total marrow population. (E) Next-generation sequencing analysis of variant allele fraction for clones and subclones in this same CMML patient at time of diagnosis, after therapy with a hypomethylating agent (HMA), and after nivolumab therapy. The level of donor single nucleotide variants post PBSCT and nivolumab therapy is depicted in orange.
Assessment of immunologic correlative studies. (A) Time course of immune cell populations at baseline and on treatment through cycle 8 (C8), including total T cell (CD3+), CD4+, and CD8+ T cell, B cell (CD20+), NK cell (CD56+CD3−), and DC (HLA-DR+LINEAGE−). Each dot represents median value. *P ≤ .05 compared with the baseline value. (B) Baseline %PD-1 expression in T-cell subsets in patients who went on to achieve response (CR/PR; green), SD (yellow), or progressive disease (PD) (orange) as best response. Red color represents the maximum value, and the blue color represents the minimum value in each row. (C) CD4 and CD8 T-cell subsets at cycle 4 according to response status, including Treg and Tcon. Red color represents the maximum value, and the blue color represents the minimum value in each row. (D) % of morphologic and cytogenetic bone marrow involvement as well as immune activity from CD3+CD8+ T-cell and donor-cell chimerism in a responding patient with CMML at baseline (green), day +75 post nivolumab (red), and day +112 post nivolumab (blue). The % of trisomy 21 is 100% of 10 XY recipient metaphase cells that were evaluated by the clinical cytogenetics laboratory, while the % of CTLs was 20% of the total marrow population. (E) Next-generation sequencing analysis of variant allele fraction for clones and subclones in this same CMML patient at time of diagnosis, after therapy with a hypomethylating agent (HMA), and after nivolumab therapy. The level of donor single nucleotide variants post PBSCT and nivolumab therapy is depicted in orange.
Discussion
We previously found that CTLA-4 blockade could augment GVT and effectively treat relapsed HMs after alloHCT.9 PD-1 blockade is known to be active in certain lymphoid malignancies, but in retrospective studies was found to increase the risk and severity of GVHD post–alloHCT.15,16 Here, we report the first prospective trial of nivolumab for post–alloHCT relapse of HMs, and we identify a low dose of 0.5 mg/kg as the MTD.
Stimulation of graft immunity through PD-1 blockade carries a risk of inducing irAEs and GVHD. In our study, despite using low doses of nivolumab, 39% of patients developed new or worsening a/c GVHD, which was fatal in 2 patients. Both the high rate of irAEs and the challenge of managing such toxicities is similar to the retrospective series.15,16 Our prior data with CTLA-4 blockade demonstrated less frequent rates of GVHD, with 14% of patients experiencing GVHD that precluded further administration of ipilimumab.9 Interestingly, this difference between CTLA-4 vs PD-1 blockade was also observed previously in earlier preclinical studies in murine models, where more potent acceleration of GVHD lethality was seen after PD-1 blockade relative to CTLA-4 blockade.10,11 In these models, administration of anti-PD1 antibodies at earlier time points was associated with more severe donor T-cell GVH responses.10 We similarly observed that a shorter interval between transplantation and first nivolumab infusion was associated with higher risk of developing GVHD. Our study included only patients who had undergone transplant ∼6 or more months prior to enrollment. It is possible that using nivolumab later post–alloHCT or in even lower doses could be less toxic, although the inability to use it early after alloHCT may limit the eligible population, since many patients relapse within a few months of transplantation.1
Our study population was heterogeneous, which allowed us to look for toxicity patterns and an efficacy signal across a range of different diseases, although also limited our ability to provide more definitive efficacy data for any one particular disease. Modest antitumor activity was observed, with an ORR of 32%, with several responses seen in patients with lymphoid malignancies (ORR 44%) known to be sensitive to PD-1 blockade outside of the alloHCT setting and less activity in patients with myeloid malignancies (ORR 21%). In contrast to our prior experience treating patients with ipilimumab in a similar population,9 none of the 6 patients with extramedullary relapse of myeloid disease responded to nivolumab. Although the 1-year PFS was low at 23%, the higher 1-year OS rate of 56% raises the question of whether nivolumab might lead to greater activity of the next line of therapy, as has been suggested in HL26 ; however, this cannot easily be assessed in the present trial, as data on subsequent therapies were not routinely collected. Given the paucity of effective alternatives for patients who relapse after alloHCT, further investigation of PD-1 blockade for patients with diseases known to be sensitive to PD-1 blockade outside the alloHCT setting (eg, HL, PMBCL) may still be warranted particularly with a focus on novel strategies to mitigate toxicities. Conversely, the low level of efficacy outside of those histologies, and in myeloid malignancies in particular, together with the significant risk of toxicities, may justify caution against further attempts to study PD-1 blockade in these populations.
Our immune correlative analyses provide some initial insights that may help inform the design of future trials of PD-1 blockade in patients with HL who relapse post–alloHCT. We found that responders or patients with SD had higher baseline expression of PD-1 in T-cell subpopulations in the blood, including CD4+ and CD8+ cells. Patients with early progression of disease had a higher Treg/Tcon ratio, suggesting less robust antitumor activity. Interestingly, we found that response in a patient with a myeloid malignancy was preceded by a polyclonal increase in CTLs, along with increased donor cell chimerism, and that genetic subclones had differential sensitivity to therapy. This suggests a potential way to identify patients who may benefit from combination partners with anti–PD-1 therapy. If similar biomarkers of tolerability can be identified, these hypothesis-generating findings could warrant further investigation in selected patients.
Alternative ways to safely promote GVT in this setting could leverage the possible benefit of HMAs. One such approach combines donor lymphocyte infusion with HMAs and has demonstrated efficacy particularly in patients with molecular and/or late recurrences in myeloid diseases.27,28 In addition, a recent phase 1 trial for relapsed MDS/AML after alloHCT found that sequential administration of lenalidomide with azacitidine was effective and tolerable, with no GVHD-related mortality.29 Interestingly, mouse models have shown that in vivo pretreatment with HMAs may mitigate GVHD via upregulation of Treg, without negatively impacting the GVT effect.30 Based on our prior experience with CTLA-4 blockade in post–alloHCT relapse of myeloid malignancies, we are pursuing additional trials to identify optimal combination partners for ipilimumab, including an ongoing study of decitabine plus ipilimumab in patients with relapsed MDS/AML post–alloHCT (#NCT02890329).31 A recent study also suggests the potential of HMAs to augment the antitumor activity and possibly mitigate immune-mediated toxicities of PD-1 blockade in HL,32 a strategy that could be worthy of further exploration for HL patients who relapse post–alloHCT.
In summary, our early phase study defined nivolumab 0.5 mg/kg as the MTD in the post–alloHCT population, although even at this low dose significant immune-mediated toxicities were observed. This dose is substantially lower than is typically used outside the allo transplant setting, which has important and immediate implications for those considering off-label use of nivolumab in the post–alloHCT population in clinical practice. Minimal antitumor activity was observed in myeloid malignancies, and several immune toxicities occurred. Encouraging activity was observed in patients with HL. Further investigation of PD-1 blockade post–alloHCT in this population could evaluate biomarker-based strategies to mitigate toxicity and direct this therapy to those most likely to derive clinical benefit.
Deidentified individual participant data that underlie the reported results will be made available 6 months after publication for a period of 5 years after the publication date through CTEP. Proposals for access should be sent to matthew_davids@dfci.harvard.edu. The study protocol is included as a data supplement available with the online version of this article.
Presented in part at the 2018 annual meeting of the American Society of Hematology in San Diego, CA, 3 December 2018.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients and their families who participated in this trial; the research coordinators, research nurses, advanced practice providers, and site staff for their support of the trial; Carol Reynolds for flow cytometry; and Irene Ghobrial, Lee Greenberger, and Michael Yaffe for support of the trial through the Leukemia & Lymphoma Society (LLS) BCRP.
This study was supported by research funding from the National Institutes of Health, National Cancer Institute grants 5R01CA183559, 5R01CA183560, and 5UM1CA186709 (Principal Investigator: Geoffrey Shapiro), National Cancer Institute CTEP, Bristol-Myers Squibb, LLS Therapy Accelerator Program, Pasquarello Tissue Bank, DFCI Department of Medical Oncology, and Center for Immuno-Oncology. M.S.D. and P.A. are both LLS Scholars in Clinical Research.
Authorship
Contribution: M.S.D., H.T.K., H.S., R.J.S., and P.A. conceived and designed the study; M.S.D., C. Costello, A.F.H., F.L.L., R.O.M., A.S., M.M., A.W., D.A., Y.-B.C., S.N., V.T.H., C. Cutler, E.P.A., P.B., E.D.B., A.B., R.J.S., and P.A. acquired the data; M.S.D., H.T.K., R.J.S., and P.A. analyzed and interpreted the data; H.T.K., A.W., P.B., C.J.W., and J.R. provided correlative analyses; M.S.D., A.A., A.P.B., R.J.S., and P.A. wrote the paper; and H.T.K. performed the statistical analysis.
Conflict-of-interest disclosure: M.S.D. received an institutional research grant from Bristol-Myers Squibb during the conduct of this study, as well as personal fees from AbbVie, Acerta Pharma, Adaptive Biotechnologies, Ascentage, AstraZeneca, Beigene, Celgene, Genentech, Gilead Sciences, InCyte, Infinity Pharmaceuticals, Janssen, MEI Pharma, Merck, Pharmacyclics, Research to Practice, Roche, Syros Pharmaceuticals, TG Therapeutics, and Verastem, and institutional research funding from Acerta Pharma, Ascentage, Genentech, Infinity Pharmaceuticals, MEI Pharma, Pharmacyclics, Surface Oncology, TG Therapeutics, and Verastem, outside the submitted work. A.F.H. has received personal fees from BMS, Genentech, Merck, Kite, Adaptive, Seattle Genetics, and Gilead, as well as institutional research support from Bristol-Myers Squibb, Genentech, Immune Design, AstraZeneca, Merck, Seattle Genetics, Kite Pharma, and Gilead outside the submitted work. F.L.L. has received personal fees from Kite Pharma, Novartis, GammaDelta T cell Therapeutics, and Cellular BioMedicine Group Inc outside the submitted work. D.A. has received personal fees from BMS, Celgene, Juno, Partners Tx, Karyopharm, Aviv MedTech Ltd, Janssen, Parexel, Takeda, as well as institutional research support from Celgene and Pharmacyclics, outside the submitted work. Y.-B.C. has received personal fees from Incyte, Kiadis, Magenta, Takeda, as well as institutional research support from AbbVie, outside the submitted work. V.T.H. has received personal fees from Jazz Pharmaceuticals, Omeros, and Alexion, outside the submitted work. C.J.W. is a cofounder of Neon Therapeutics and has received institutional research funding from Pharmacyclics, outside the submitted work. J.R. reports institutional research funding from Equillium and Kite Pharma and personal fees from Aleta Biotherapeutics, Avrobio, Celgene, Draper Labs, LifeVault Bio, Talaris Therapeutics, and TScan Therapeutics, outside the submitted work. R.J.S. reports personal fees from Kiadis, Juno, Gilead, Neovii, Cugene, Jazz Pharmaceuticals, and Mana Therapeutics, outside the submitted work. P.A. has received personal fees from Bristol-Myers Squibb, Merck, Pfizer, Affimed, Adaptive, Infinity, ADC Therapeutics, and institutional research funding from Bristol-Myers Squibb, Merck, Affimed, Adaptive, Roche, Tensha, Otsuka, Sigma τ, Genentech, and Kite, outside the submitted work. The remaining authors declare no competing financial interests.
A list of the Leukemia & Lymphoma Society Blood Cancer Research Partnership sites that participated in the trial appears in the supplemental appendix.
Correspondence: Matthew S. Davids, Dana-Farber Cancer Institute, Department of Medical Oncology, 450 Brookline Ave, Boston, MA 02215; e-mail: matthew_davids@dfci.harvard.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal