

In this issue of Blood, have performed and integrated a comprehensive multiomics (genome, epigenome, and transcriptome) analysis of patients who have undergone blastic transformation of chronic myeloid leukemia (CML).1
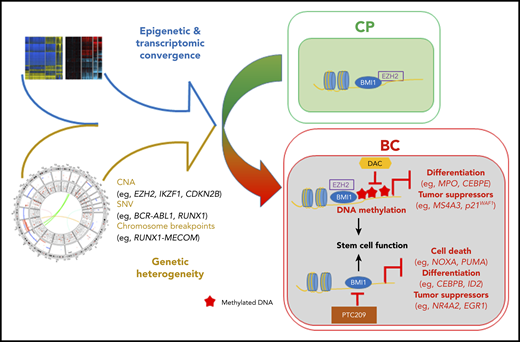
Relatively heterogeneous mutations and the epigenetic state converge to generate a convergent transcriptional program in the transition from CML CP to BC. This convergent state is driven, in part, by interplay between the PRC and DNA methylation and can potentially be antagonized by combinatorial inhibition. CNA, copy number alteration; SNV, single-nucleotide variant. See Figure 7C in the article by Ko et al that begins on page 2337.
Relatively heterogeneous mutations and the epigenetic state converge to generate a convergent transcriptional program in the transition from CML CP to BC. This convergent state is driven, in part, by interplay between the PRC and DNA methylation and can potentially be antagonized by combinatorial inhibition. CNA, copy number alteration; SNV, single-nucleotide variant. See Figure 7C in the article by Ko et al that begins on page 2337.
CML is initiated, and subsequently driven by, a specific chromosomal translocation, the Philadelphia chromosome, rearranging the BCR and ABL genes to encode the BCR-ABL fusion protein, a constitutively activated protein tyrosine kinase.2 This results in a chronic myeloproliferative neoplasm characterized by an overproduction of granulocytes and their progenitors.2 If left untreated, CML will invariably and inexorably progress from chronic phase (CP) to an aggressive and often fatal acutelike leukemia: myeloid or lymphoid blast crisis (MBC or LBC, respectively). Despite the transformative introduction and subsequent evolution of ABL-selective tyrosine kinase inhibitors (TKIs) >20 years ago, CML still remains a story of successful management, rather than eradication of disease. The majority of patients diagnosed in CP respond well to treatment, with 10-year survival rates of >80% in some selected series,3 although less impressive results are seen in real-world studies. However, only ∼10% of these patients will remain in therapy-free remission upon TKI discontinuation4 ; hence, a major effect of TKI appears to be their ability to alter the natural history of progression to blast crisis (BC) through BCR-ABL inhibition and leukemic bulk reduction.
Despite the dismal outcomes seen in BC-CML, the mechanisms and drivers of progression remain poorly understood. This reflects its relative rarity in the post-TKI era, but also the lack of faithful models in which to derive mechanism and generate and test hypotheses. Mouse models of BCR-ABL disease exist, but often only model CP5 or present with LBC6 ; however, a recent model did replicate disease progression and heterogeneity and identified potential therapeutic targets.7 Similarly, human studies are sparse; genetic studies have generally involved small patient cohorts and have focused on detecting mutations in single genes. Gene expression studies, such as the seminal work by Radich et al,8 have been more informative and involved larger numbers of patients; however, novel therapeutic strategies are still lacking. This report from Ko et al addresses many of these deficiencies. This study provides a much needed and, given the rarity of the disease, large (∼74 patients) data set, of CP and BC-CML patient samples, with many paired.1 For these samples, the mutational landscape, as defined by whole-genome and -exome sequencing, was integrated with gene expression by array technology, methylome profiling using arrays, and chromatin immunoprecipitation followed by high-throughput sequencing to identify patterns and regulators of BC transformation (see figure).
Regarding the mutational landscape, the genotoxic effects of BCR-ABL are well described experimentally; therefore, it is of great interest that the authors demonstrate only a modest mutational burden for both MBC and LBC, comparable to other acute leukemias. Of note, a mutational pattern that appeared specific for BRC-ABL was detected but, again rather surprisingly, this pattern was independent of ROS or APOBEC signatures. Taken together, these findings suggest little clinical evidence for significant genotoxicity for BCR-ABL activity in vivo. The mutational findings in general confirmed previous analyses; however, given the heterogeneity of these mutations, perhaps one of the most surprising findings of this study is the highly congruent transcriptomes of individual CML-BC cases. This finding even extended to the different lineages, where, despite being enriched for gene signatures found in their normal (and malignant) counterparts (ie, acute myeloid leukemia and acute lymphoblastic leukemia), MBC and LBC CD34+ blasts exhibited highly similar core signatures, with inflammation and quiescence as central themes. This finding may suggest a common mechanism of transformation, perhaps related to the permissive or selective effect of BCR-ABL signaling, as BCR-ABL mutations and therefore continued activity were very frequent, or potentially to the transformation of the same or a similar cell of origin. Of potential clinical importance, it also suggests that therapeutically targeting these pathways may be efficacious in both MBC and LBC.
The authors also show enrichment of polycomb repressive complex (PRC)-related gene mutations as a complementation group associated with CML progression. Integrating these findings with their other epigenetic layers, they propose the PRC1 (BMI1) and PRC2 (EZH2) complexes as potential mediators of transformation through an epigenetic reprograming of the CML-BC core transcriptional signature. The PRC2 complex and its catalytic component EZH2 have already been associated with CML-BC9,10 ; however, description of other recurrent PRC-component alterations is novel and may have therapeutic implications. Indeed, the authors attempted to deconvolute the individual effects of the PRC1 and PRC2 complexes on CML-BC maintenance, using clinical-grade inhibitors of PRC1 and PRC2 alone and in combination with hypo- (DNA) methylating agents (HMAs) that are already in widespread clinical usage. The interesting therapeutic combination of a PRC1 inhibitor and an HMA showed promising results in their in vitro systems, and this promise should be tested in vivo in future studies.
However, perhaps the most interesting findings of this study lie within the apparent different functions of the PRC1 and PRC2 complexes and the proposed epigenetic crosstalk between PCR2/EZH2 and DNA methylation during progression to BC. Although BMI1/PRC1 appears to maintain the established BC transcriptional signature, PRC2/EZH2 function appears to “prime” genes for subsequent DNA methylation–specific repression. In addition, by demonstrating that genes prognostic for/implicated in transformation are more likely to be PRC-bound during the CP and methylation-prone in BC, the authors also raise the intriguing possibility of reverting poor prognostic patients to a lower-risk category by utilizing clinically available epigenetic therapies. It remains to be seen if the epigenetic deregulation already evident in CP and that evolves during BC progression is somehow linked to, or directly induced through, the activity of BCR-ABL and its downstream signaling cascades to chromatin. This is an obvious question that merits further investigation, particularly in the current era of single-cell technologies.
Like all good studies, this analysis asks as many questions as it answers. Does the convergent pattern of gene expression reflect a common mechanism of transformation, or is it merely a common effector of multiple mechanisms? What is the relationship of this common pathway to BCR-ABL signaling? Are PRC complexes and the DNA methylation machinery viable targets in CML-BC? As thorough and detailed as this report may be, these results might have only scratched the surface of the information that these data sets hold. Further study is therefore warranted, and this invaluable data set will provide a rich resource to facilitate such studies.
Conflict-of-interest disclosure: G.G. and B.J.P.H. declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal