

Key Points
The benefit of GO in non-CBF AML patients with favorable or intermediate ELN 2017 risk is restricted to those with signaling mutations.
Higher CD33 expression levels on non-CBF AML blasts correlate with the presence of activating signaling mutations.

Abstract
Acute myeloid leukemia (AML) is a highly heterogeneous disease both in terms of genetic background and response to chemotherapy. Although molecular aberrations are routinely used to stratify AML patients into prognostic subgroups when receiving standard chemotherapy, the predictive value of the genetic background and co-occurring mutations remains to be assessed when using newly approved antileukemic drugs. In the present study, we retrospectively addressed the question of the predictive value of molecular events on the benefit of the addition of gemtuzumab ozogamicin (GO) to standard front-line chemotherapy. Using the more recent European LeukemiaNet (ELN) 2017 risk classification, we confirmed that the benefit of GO was restricted to the favorable (hazard ratio [HR], 0.54, 95% confidence interval [CI], 0.30-0.98) and intermediate (HR, 0.57; 95% CI, 0.33-1.00) risk categories, whereas it did not influence the outcome of patients within the adverse risk subgroup (HR, 0.93; 95% CI, 0.61-1.43). Interestingly, the benefit of GO was significant for patients with activating signaling mutations (HR, 0.43; 95% CI, 0.28-0.65), which correlated with higher CD33 expression levels. These results suggest that molecular aberrations could be critical for future differentially tailored treatments based on integrated genetic profiles that are able to predict the benefit of GO on outcome.
Introduction
Major advances were made over the past decades in defining underlying cytogenetic and molecular aberrations routinely used to inform acute myeloid leukemia (AML) classifications and stratify patients into prognostic subgroups.1 Although the prognostic relevance of molecular alterations has been well investigated, translation of this information into clinical care is ongoing.2 Until recently, standard induction chemotherapy in AML patients relied mostly on the “3+7” regimen combining an anthracycline plus cytarabine. However, recent studies have highlighted newly approved medications directed against AML-specific antigens or pathways.3 Given this game-changing context, there is a need to analyze the impact of these new drugs in light of the complex AML molecular landscape.
Here, we report the extensive mutational analysis of a well-annotated cohort of AML patients aged 50 to 70 years old. Patients were randomized into the Acute Leukemia French Association (ALFA)-0701 trial (ClinicalTrials.gov Identifier: NCT00927498),4 which evaluated the added value of fractionated doses of gemtuzumab ozogamicin (GO) to standard front-line chemotherapy.3 Results from our group4 and other investigators5-7 suggested that the addition of GO to chemotherapy might result in a substantial improvement in patient outcome, especially in AML patients with nonadverse cytogenetics8 and/or high CD33 expression.9,10 Considering these findings and the widespread use of high-throughput sequencing technologies in current clinical care, we retrospectively addressed the question of the predictive value of molecular events on the benefit of GO in the ALFA-0701 study.
Study design
A total of 235 patients with non–core binding factor (CBF) AML was studied for molecular aberrations using a 43-gene targeted panel (supplemental Methods, available on the Blood Web site). FLT3-internal tandem duplication (ITD) screening was performed by fragment analysis, as previously described,8 and the allelic ratio (AR) was determined as the ratio of the area under the curve FLT3-ITD/FLT3 wild type. All samples were screened for recurrent gene rearrangements and KMT2A–partial tandem duplication using reverse transcriptase multiplex ligation-dependent probe amplification assay.11 CD33 expression was evaluated as previously described.9 Single-gene analyses were restricted to subgroups with >15 patients. The “signaling mutations” group was defined by the presence of ≥1 mutation in FLT3, NRAS, KRAS, PTPN11, JAK2, RIT1, or CBL, regardless of the variant allelic frequency (VAF).
Results and discussion
Mutational profiling
A total of 221 patients (94%) had ≥1 mutation. Eleven genes were recurrently mutated in ≥10% of patients: NPM1 (34%), DNMT3A (29%), FLT3 (28% [FLT3-ITD 21%; FLT3-TKD 8%]), TET2 (20%), RUNX1 (16%), NRAS (14%), IDH2 (13%), SRSF2 (13%), ASXL1 (13%), TP53 (11%), and IDH1 (11%) (supplemental Figures 1 and 2). According to the European LeukemiaNet (ELN) 2017 risk classification, 71 (30%), 69 (29%), and 95 (40%) patients had favorable, intermediate, and adverse risk, respectively. The distributions of gene mutations, ELN risk categories,1 and CD33 expression9 were well balanced between the control group and the GO group (supplemental Table 1).
Benefit of GO in molecularly defined subgroups
The global complete remission (CR) rate with or without platelet recovery was 71% (166/235), with no difference between the GO and control arms. The CR rate was 86%, 83%, and 64% in the ELN favorable-risk, intermediate-risk, and adverse-risk categories, respectively. None of the ELN risk subgroups or gene mutations benefited from GO in terms of CR rate (data not shown).
For the 235 patients, the 3-year event-free survival (EFS) was 25.7% (95% confidence interval [CI], 2.6-30.0). The benefit of GO was restricted to the ELN favorable-risk (hazard ratio [HR], 0.54; 95% CI, 0.30-0.98) and intermediate-risk (HR, 0.57; 95% CI, 0.33-1.00) categories, whereas it did not influence the outcome of patients with adverse risk (HR, 0.93; 95% CI, 0.61-1.43) (Figure 1A-C). At the molecular level, a benefit of GO was observed in patients with NPM1 (HR, 0.48; 95% CI, 0.28-0.83), FLT3-ITD (HR, 0.36; 95% CI, 0.18-0.72), FLT3-TKD (HR, 0.20; 95% CI, 0.05-0.78), NRAS (HR, 0.43; 95% CI, 0.19-0.95), KRAS (HR, 0.27; 95% CI, 0.09-0.84), and SRSF2 mutations (HR, 0.28; 95% CI, 0.12-0.65) (Figure 2A; supplemental Table 2; supplemental Figure 3). In contrast, no benefit was observed in patients with adverse-risk molecular factors1 RUNX1, ASXL1, and TP53. Considering mutational classes12 (supplemental Methods), a greater benefit of GO was observed in patients with activating signaling mutations (HR, 0.43; 95% CI, 0.28-0.65) or epigenetic (HR, 0.68; 95% CI, 0.48-0.97) or spliceosome deregulation (HR, 0.44; 95% CI, 0.24-0.80). The predominant benefit of signaling mutations led us to assess the interaction between these mutations and GO in ELN 2017 favorable/intermediate-risk patients (Figure 1D-E). With regard to EFS and overall survival (OS), patients with signaling mutations benefited from GO (EFS: HR, 0.36; 95% CI, 0.21-0.61 and OS: HR, 0.47; 95% CI, 0.26-0.83), whereas patients without signaling mutations did not (EFS: HR, 1.07; 95% CI, 0.53-2.16 and OS: HR, 1.11; 95% CI, 0.50-2.47). The interaction between a benefit of GO and the presence of a signaling mutation was significant for EFS (P = .02), with only a trend for OS (P = .07). Because many cooperating mutations may be associated in the same patients, we next assessed whether the benefit of GO observed in patients with NPM1, epigenetic, or spliceosome mutations was restricted to those patients with signaling mutations. In these 3 subgroups, a significant benefit in terms of EFS was only observed in patients with co-occurring signaling mutations (supplemental Figure 4). Interaction was significant for patients with NPM1 or epigenetic mutations. Altogether, these observations suggested that a benefit of GO was restricted to patients with activating signaling mutations.
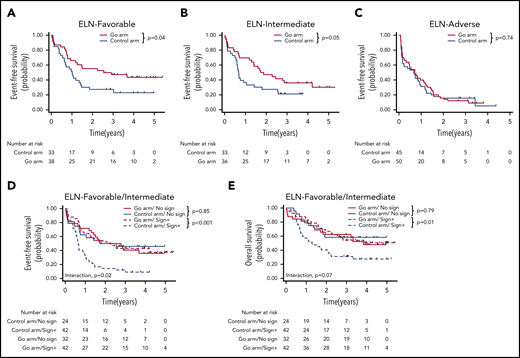
Benefit of GO according to ELN classification and the existence of signaling mutation. (A-C) EFS according to ELN 2017 subgroups. EFS (D) and OS (E) according to protocol arm and the existence of signaling mutations.
Benefit of GO according to ELN classification and the existence of signaling mutation. (A-C) EFS according to ELN 2017 subgroups. EFS (D) and OS (E) according to protocol arm and the existence of signaling mutations.
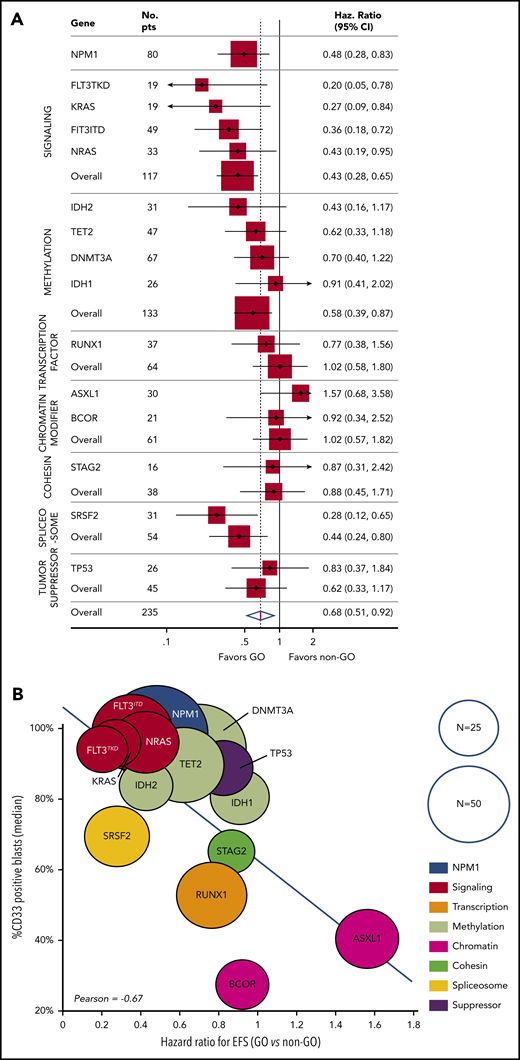
Benefit of GO according to mutation profile and correlation with CD33 expression. (A) The forest plot shows the HR for EFS in the GO arm vs the control arm for each mutation (with N ≥ 15) and mutation subgroup. (B) The correlation between the HRs and CD33 expression on AML blasts. Each circle represents a subgroup of patients with a mutation. The size of each circle is proportional to the number of patients.
Benefit of GO according to mutation profile and correlation with CD33 expression. (A) The forest plot shows the HR for EFS in the GO arm vs the control arm for each mutation (with N ≥ 15) and mutation subgroup. (B) The correlation between the HRs and CD33 expression on AML blasts. Each circle represents a subgroup of patients with a mutation. The size of each circle is proportional to the number of patients.
Finally, we investigated the impact of FLT3-ITD AR on GO benefit. An AR cutoff was defined as the median value of 0.4 (supplemental Figure 5). In patients with FLT3-ITD, there was no difference in HR (GO vs control) for EFS between the high-AR group (HR, 0.42; 95% CI, 0.16-1.08) and the low-AR group (HR, 0.35; 95% CI, 0.12-0.99). We then considered a composite VAF for signaling mutations as the sum of individual gene VAFs. A VAF cutoff was defined as the median value of 25%. A slightly lower HR for EFS was observed in patients with high VAF (HR, 0.36; 95% CI, 0.19-0.66) compared with patients with low VAF (HR, 0.50; 95% CI, 0.28-0.92). However, both profiles led to a benefit of GO in terms of EFS.
Correlation between benefit of GO and CD33 expression
A correlation between GO activity and CD33 target expression was suggested by our group9 and other investigators.10 To further explore the association between signaling mutations and benefit of GO, we compared the presence of mutations with CD33 expression levels on AML blasts. Interestingly, the benefit of GO in terms of EFS was correlated with CD33 expression levels among the different gene mutations, with particularly high levels observed on signaling mutation-positive AML blasts (Pearson correlation coefficient, −0.67; Figure 2B). Among patients with epigenetic mutations, the level of CD33 expression was significantly higher in patients with signaling mutations (98% vs 60%; P < .001). This difference was not observed in patients with NPM1 or spliceosome mutations.
A biological mechanism linking CD33 expression and signaling mutations is still missing. Such molecular events co-occur in a wide range of hematological malignancies and are generally considered secondary or late clonal events.13 Signaling mutations are found in up to 75% of patients with CBF AML,13 a group for which a clear benefit of GO was demonstrated,5 despite a characteristically low expression of CD33.10 With no evidence of direct control of activating signaling on CD33 expression, one could hypothesize that the interaction observed could reflect leukemic cell adaptation to its microenvironment. CD33 belongs to the Siglec family and is a transmembrane glycoprotein receptor committed to myeloid lineage. CD33 harbors 2 tyrosine residues on its cytoplasmic domain that function as docking sites for the tyrosine phosphatases SHP-1 and SHP-2.14 Thus, CD33 is considered an inhibitory receptor that negatively regulates cell proliferation and activation.15-17 In this context, activated kinase signaling could be a coevolutionary pattern that overcomes CD33 downstream effects on leukemic cell development.
In conclusion, in the context of the ALFA-0701 trial, we showed that the benefit of GO in patients is determined by underlying molecular aberrations and is primarily restricted to patients bearing signaling mutations associated with high CD33 expression. This observation could be critical for future differentially tailored treatments based on integrated genetic profiles. Also, future investigations should evaluate the relevance of the use of GO considering other targeted therapy, including FLT3 inhibitors.
All requests for sequence data, BAM files, and VCF files should be sent to Nicolas Boissel (nicolas.boissel@aphp.fr).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Christophe Roumier and the Tumor Bank for the ALFA group (Lille University Hospital, certification NF 96900-2014/65453-1) for handling, conditioning, and storing patient samples. The work of all physicians and clinical research assistants is also acknowledged.
Authorship
Contribution: E.F., N.D., B.D., A.M.-R., M.C., and C. Preudhomme performed molecular analyses; E.G. reviewed flow cytometry data; C.T. reviewed cytogenetics; E.R, P.T., D.C., X.T., S. Chantepie, J.-V.M., E.L., K.C.-L., J.L., C. Pautas, H.D., S. Castaigne, C. Preudhomme, and N.B. contributed to the execution of the study and collected and assembled the data; and E.F., N.D., and N.B. wrote the manuscript, which was approved by all of the authors.
Conflict-of interest disclosure: S. Castaigne has received consultancy fees from Pfizer, and H.D. has received honoraria from Pfizer for serving on advisory boards. The remaining authors declare no competing financial interests.
A complete list of the members of the Acute Leukemia French Association appears in “Appendix.”
Correspondence: Nicolas Boissel, Hematology Department, Saint-Louis Hospital, 1 Avenue Claude Vellefaux, 75010 Paris, France; e-mail: nicolas.boissel@aphp.fr.
Appendix: study group members
The members of the Acute Leukemia French Association are J. P. Marolleau, G. L. Damaj, B. Royer, B. Gruson, A. Charbonnier, H. Copin, J. F. Claisse, A. Parcellier, A. Aleme (CHU, Amiens); L. Sutton, A. Al Jijakli, T. H. Mossafa, H. Delaouni, Guerekobaya (CH, Argenteuil); C. Gardin, T. Braun, L. Ades, P. Fenaux, D. Lusina, V. Eclache, H. Chehimi, S. Pineau (Hôpital Avicenne, Bobigny); B. Choufi, C. Roche-Lestienne, M. Brument (CH, Boulogne/Mer); O. Reman, M. Macro, S. Chantepie, S. Cheze, H. Johnson Ansah, K. Benabed, D. Naguib, M. Decamp, M. Malet, X. Troussard, E. Marin (CHU, Caen); J. V. Malfuson, J. Konopacki, T. de Revel, T. Fagot, H. Mossafa, D. Bories, V. Foissaud (Hôpital Percy, Clamart); C. Salanoubat, S. Haiat, A. Devidas, B. Joly, S. Bouledroua, C. Petitdidier, S. Cereja, H. Mossafa, I. Lemaire, P. Quillet (CH, Corbeil); C. Pautas, C. Cordonnier, S. Maury, A. Toma, Y. Hicheri, D. Bories, O. Wagner-Ballon, C. Perot, L. Cabanne (Hôpital Henri Mondor, Créteil); J.N. Bastie, E. Ferrant, O. Casasnovas, D. Caillot, I. Lafon, E. Ferrant, A. Rascalou-Waultier, F. Mugneret, M. Maynadier, J. R. Tessier, C. Lagouge, D. Ragonneau (CHU, Dijon); M. Bemba, F. Bonnevie, M. Wetterwald, C. Delattre, C. Roche-Lestienne, V. Paquez (CH, Dunkerque); S. de Botton, J. H. Bourhis, J. B. Micol, N. Auger, V. Saada (Institut Gustave Roussy, Villejuif); B. Dupriez, L. Stalnikiewicz, H. Declercq, H. Vandeputte, A. Daudignon (CH, Lens); B. Quesnel, C. Berthon, C. Roche-Lestienne, A. Renneville, C. Preudhomme, O. Nibourel, C. Rodriguez, C. Frimat, H. Debarri, H. Djeda (CHU, Lille); P. Turlure, S. Girault, L. Remenieras, D. Bordessoule, M. P. Gourin, A. Jaccard, S. Moreau, N. Gachard, F. Trimoreau, J. Abraham, M. Touati, C. Philippon (CHU, Limoges); X. Thomas, F. Nicolini, J. Troncy, E. Wattel, H. Labussière, M. Michallet, S. Hayette, A. Plesa, I. Tigaud, S. Ducastelle. F. Barraco, M. Elhamri (Hôpital Lyon Sud, Lyon); J. Frayfer, W. Abarah, A. Cung, H. Mossafa, H. Akioud (CH, Meaux); L. Gastaud, A. Thyss, S. Raynaud, C. Debaigt (Centre Lacassagne, Nice); L. Manonne, F. Legrand, I. Sudaka, S. Raynaud, C. Hathroubi (CHU, Nice); O. Hermine, A. Marçais, F. Suarez, R. Delarue, F. Sicre de Fonbrune, D. Sibon, A Chabanon, L. Frenzel, I. Radfortd-Weiss, C. Brouzes, T. Saheb (Hôpital Necker, Paris); M. Uzunov, S. Nguyen, V. Leblond, F. Nguyen-Khac, F. Davy, K. Maloum, G. Candellier (Hôpital Pitié Salpétrière, Paris); H. Dombret E. Raffoux, N. Boissel, E. Lengliné, J. B. Micol, T. Cluzeau, J. M. Miclea, A. de Labarthe, O. Maarek, J. M. Cayuela, A. Andreoli, K. Celli-Lebras, R. Grapin, F. Hilaire, A. S. Debrie, (Hôpital Saint-Louis, Paris); L. Fouillard, I. Vaida, H. Gonzalez, R. Benramdane, D. Mallet, C. Terré, M. Amirault (CH, Pontoise); I. Plantier, L. Detourmignies, I. Sendrier Dervite, C. Ghevaert, K. Dernivoix (CH, Roubaix); H. Tilly, E. Lemasle, N. Contentin, A. Stamatoulas, F. Jardin, S. Lepretre, P. Lenain, C. Bastard, M. P. Callat, P. Etancelin, D. Penther, S. Vaudaux (Centre Henri Becquerel, Rouen); M. Janvier, J. Vargaftig, S. Glaisner, C. Soussain, C. Terré, H. Bennani, P. Vavasseur, (Centre Rene Huguenin, Saint-Cloud); S. Castaigne, P. Rousselot, H. Farhat, S. Ghez, J. Lambert, S. Rigaudeau, A. L. Taksin, C. Terré, I. Garcia, S. Raggueneau, C. Leterme (CH, Versailles); M. Simon, J. Fernandes, A. Daudignon, H. Bisiau, C. Bremeault (CH, Valenciennes).

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal