


Abstract
Secondary acute myeloid leukemias (AMLs) evolving from an antecedent myeloproliferative neoplasm (MPN) are characterized by a unique set of cytogenetic and molecular features distinct from de novo AML. Given the high frequency of poor-risk cytogenetic and molecular features, malignant clones are frequently insensitive to traditional AML chemotherapeutic agents. Allogeneic stem cell transplant, the only treatment modality shown to have any beneficial long-term outcome, is often not possible given the advanced age of patients at time of diagnosis and frequent presence of competing comorbidities. Even in this setting, relapse rates remain high. As a result, outcomes are generally poor and there remains a significant unmet need for novel therapeutic strategies. Although advances in cancer genomics have dramatically enhanced our understanding of the molecular events governing clonal evolution in MPNs, the cell-intrinsic and -extrinsic mechanisms driving leukemic transformation at this level remain poorly understood. Here, we review known risk factors for the development of leukemic transformation in MPNs, recent progress made in our understanding of the molecular features associated with leukemic transformation, current treatment strategies, and emerging therapeutic options for this high-risk myeloid malignancy.
Introduction
The Philadelphia-negative myeloproliferative neoplasms (MPNs) comprise a spectrum of clonal hematopoietic disorders characterized by aberrant proliferation of mature myeloid elements manifesting as an excess of red blood cells (polycythemia vera [PV]), platelets (essential thrombocytosis [ET]), and/or fibrosis-producing white blood cells (primary myelofibrosis [PMF]).1 Patients often present with constitutional symptoms and splenomegaly and are at increased risk of thrombotic and/or bleeding complications. Activating mutations of the JAK/STAT pathway, primarily JAK2, MPL, and CALR, are present in the great majority of patients at diagnosis and are vulnerable to JAK inhibition,2 underscoring the central role of this signaling pathway in the initiation and maintenance of chronic-phase MPNs.
Secondary acute myeloid leukemia (AML) arising from MPN, defined by the World Health Organization as the presence of ≥20% myeloblasts in the peripheral blood or bone marrow of a patient with an antecedent myeloproliferative disorder,3 is a well-recognized and portentous event in the natural history of an MPN and likely represents the culmination of a stepwise series of genetic and epigenetic events that engender progressive self-renewal and proliferative capacity upon an increasingly fit hematopoietic clone. “Accelerated-phase” disease, defined as a peripheral blood or bone marrow blast count of ≥10% to 19%, is perhaps emblematic of the late stages of this evolutionary process and often shares many of the high-risk features of transformed MPNs. Risk of leukemic progression varies greatly based on the presenting clinical and morphological features at time of diagnosis, with transformation risk generally highest in PMF (most studies estimate a 10-year risk of 10% to 20%), followed by PV (2% to 4%), and then ET (1%; with 2% risk at 15 years in 1 study).4-9 Although transformation to acute leukemia most commonly manifests as a myeloid phenotype, rare cases of lymphoblastic transformation have also been reported.10 Because of the frequent presence of fibrosis in patients with advanced-phase disease, core biopsy over aspirate smear is generally recommended to most adequately assess bone marrow blast count and ensure proper diagnosis.11
The molecular and morphologic features of post-MPN AML are strikingly different from those of de novo disease, suggesting a disparate path to leukemogenesis. Post-MPN AML often displays an inordinately higher rate of erythroblastic and megakaryoblastic morphology.12 The higher frequency of TP53 mutations and high-risk cytogenetic features suggest increased chromosomal instability.13,14 Furthermore, somatic alterations frequently implicated in de novo disease, including FLT3, NPM1, and DNMT3A, are frequently absent, whereas genes involved in the epigenetic regulation of DNA, including IDH1/2, TET2, ASXL1, and EZH2, as well as the spliceosome modulator SRSF2, are enriched in post-MPN AMLs,15 implying a distinct molecular pathogenesis (Figure 1).
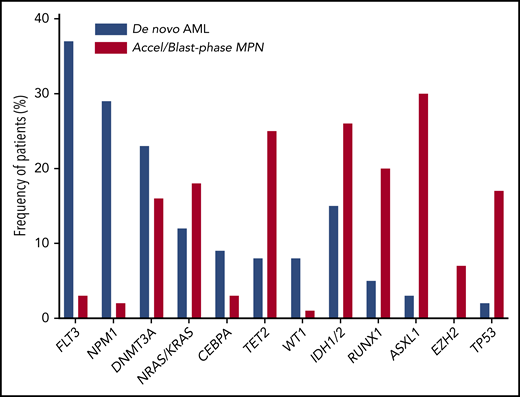
Frequency of individual somatic mutations in de novo AML vs post-MPN AML. Mutation data from a large de novo AML cohort113 in comparison to those frequently found in post-MPN AML59 reveal differences in the frequencies of commonly-mutated genes implicated in these two conditions.
The observation that post-MPN AML is pathogenetically distinct from de novo disease might explain why traditional AML therapies are so often ineffective. Clinical outcomes for patients with blast-phase MPN remain bleak, with a median survival of <6 months, and are comparatively worse than secondary AMLs arising from myelodysplastic syndrome.16,17 Furthermore, outcomes for the management of post-MPN AMLs have not changed over the last 15 years,18 underscoring the need for novel therapeutic strategies. To date, allogeneic transplant remains the only treatment modality shown to provide any long-term benefit in post-MPN AML; however, longer-term follow-up data suggest that relapse rates following transplant remain high.18,19 At the same time, newer combination strategies show some promise, and improved prognostication models incorporating an individual’s tumor molecular profile20-23 are modernizing the way clinicians are able to properly identify and treat MPN patients at increased risk of transformation.
Clinical and cytogenetic risk factors in post-MPN AML
Numerous retrospective case series have identified a recurring set of adverse clinical risk factors frequently implicated in the leukemic transformation of chronic-phase MPNs. Across multiple studies, older age (≥60-65 years), leukocytosis (≥15 × 109/L to 30 × 109/L), the presence of circulating blasts (≥3% to 10%), and worsening thrombocytopenia (<100 × 109/L) and/or anemia (with associated red blood cell transfusion need) have all consistently been shown to portend leukemic progression in MPNs (Table 1).6,8,12,24-30 It is also now well established that prior exposure to certain cytoreductive agents, including 32P, as well as pipobroman, busulfan, and other alkylating agents historically used for the treatment of PV/ET, are associated with accelerated leukemia development.7,31,32 Although some reports previously identified hydroxyurea as an independent risk factor for post-MPN AML,33,34 subsequent studies have not substantiated these claims.7,27,31,35,36 As such, hydroxyurea remains an important therapeutic consideration for symptomatic, chronic-phase disease per consensus MPN treatment guidelines.37,38 In addition, MPN subtype analyses have revealed prior thrombosis, extreme thrombocytosis, anemia, and reticulin fibrosis as factors influencing leukemic progression specifically in ET.5,39,40 Other risk factors implicated in leukemic progression across MPN subtypes for which there are conflicting or limited data include triple-negative disease (ie, negative for all known MPN drivers JAK2, MPL, or CALR),41,42 prior splenectomy,8,43-46 use of erythropoiesis-stimulating agents or danazol,28,40 and elevated serum levels of interleukin 8 (IL8)47 or C-reactive protein.48 How these additional risk factors might influence treatment decision-making at this time, however, remains unclear.
Clinical and cytogenetic/molecular risk factors for post-MPN AML
| Risk factors |
|---|
| Clinical |
| Age >60-65 y |
| Red blood cell transfusion dependency |
| Prior treatment exposure (pipobroman, 32P, chlorambucil, busulfan) |
| Prior thrombosis* |
| Myelofibrosis or prefibrotic ET/PV |
| Laboratory |
| Leukocytosis (>15 × 109/L to 30 × 109/L) |
| Anemia (Hgb <10 g/dL) |
| Thrombocytopenia (<50 × 109/L to 100 × 109/L) |
| Peripheral blast count (>1% to 10%) |
| Extreme thrombocytosis (>1000 × 109/L)* |
| Elevated serum IL-8† |
| Elevated serum C-reactive peptide† |
| Cytogenetics |
| Monosomal karyotype‡ |
| Complex karyotype or sole or 2 abnormalities that include +8, −7/7q, i(17q), −5/5q−, 12p−, inv(3), 11q23 rearrangement§ |
| Chromosome 17p deletion |
| Molecular |
| TP53, TET2, ASXL1, EZH2, SRSF2, IDH1/2, RUNX1, U2AF1Q157 |
| Risk factors |
|---|
| Clinical |
| Age >60-65 y |
| Red blood cell transfusion dependency |
| Prior treatment exposure (pipobroman, 32P, chlorambucil, busulfan) |
| Prior thrombosis* |
| Myelofibrosis or prefibrotic ET/PV |
| Laboratory |
| Leukocytosis (>15 × 109/L to 30 × 109/L) |
| Anemia (Hgb <10 g/dL) |
| Thrombocytopenia (<50 × 109/L to 100 × 109/L) |
| Peripheral blast count (>1% to 10%) |
| Extreme thrombocytosis (>1000 × 109/L)* |
| Elevated serum IL-8† |
| Elevated serum C-reactive peptide† |
| Cytogenetics |
| Monosomal karyotype‡ |
| Complex karyotype or sole or 2 abnormalities that include +8, −7/7q, i(17q), −5/5q−, 12p−, inv(3), 11q23 rearrangement§ |
| Chromosome 17p deletion |
| Molecular |
| TP53, TET2, ASXL1, EZH2, SRSF2, IDH1/2, RUNX1, U2AF1Q157 |
Hgb, hemoglobin.
Risk factors found specific to the ET subtype.
Risk factors found specific to the MF subtype.
Defined as 2+ autosomal monosomies or single autosomal monosomy associated with at least 1 structural abnormality.
As per current DIPSS-Plus criteria.24
Perhaps one of the most important predictors of leukemic progression in MPNs is chromosomal karyotyping, performed routinely in the workup of chronic-phase MPN and essential for accurate prognostication. Cytogenetic abnormalities are relatively uncommon in PV and ET at the time of diagnosis (generally <20% in PV and even rarer in ET),49,50 but are more frequent in PMF (30% to 57% of patients)4,51,52 and have been shown to accumulate over time and at blast-phase transformation.51,53,54 High-resolution single-nucleotide polymorphism array karyotyping on paired samples, for example, revealed a significant increase in the number and spectrum of genomic alterations in leukemic clones (2-3 times) in comparison with their chronic-phase counterparts (including those that transformed directly from ET or PV).14 Chromosomal alterations consistently found to portend poor-risk outcomes in MPNs include complex karyotypes (ie, any ≥3 cytogenetic changes) or any combination of ≥1 of i(17q), −5/5q−, −7/7q−, +8, 12p−, inv(3), or 11q23 rearrangements.24,52,55 In a separate study, monosomal karyotype (defined as ≥2 autosomal monosomies or 1 monosomy plus any other cytogenetic change) was particularly associated with adverse outcome, with 2-year rates of leukemic transformation as high as 29% (in comparison with 8% for complex karyotype without monosomies).56 Contemporary analyses of large patient cohorts propose a 3-tiered system over the current “favorable/unfavorable” scheme, stratifying certain lesions into a “very high-risk” category given their comparatively worse inferior survival and transformation risk.57,58 These include specifically i(17q), inv(3)/3q21, monosomy 7, 12p−/12p11.2, 11q−/11q23, and autosomal trisomies (not including +8/+9). The presence of 1 or more of these lesions corresponded with a nearly twofold rate of leukemic transformation in comparison with other traditional unfavorable karyotypes (including monosomal karyotype), and median overall survival (OS) in this patient subset was only 1.2 years,58 suggesting that the presence of any 1 or more of these specific chromosomal changes warrants special consideration for more in-depth diagnostic evaluation and therapeutic intervention.
Molecular pathogenesis and clonal evolution of post-MPN AML
It is increasingly clear that additional somatic alterations beyond typical MPN drivers (ie, JAK2V617F, CALR, and MPLW515L) occur frequently in MPN (in up to 37% to 50% of patients in some studies),20,59,60 and, like cytogenetic changes, increase in number with disease progression.61 Multiple candidate gene studies have identified a recurring set of specific mutations enriched at time of leukemic transformation, suggesting their potential role in leukemogenesis. These include, in particular, mutations of TP53, TET2, ASXL1, IDH1/2, U2AF1Q157, SRSF2, and RAS family genes,13,62-69 with TP53, RUNX1, IDH1/2, TET2, PTPN11, SRSF2, and/or ASXL1 mutations correlating independently with OS across several studies.61,62,64,66,69-72 In addition, the presence of any 2 or more somatic mutations has also been shown to predict for leukemia-free survival,4,60,73 and any 3 or more mutations has been shown to correlate with reduced response to JAK2 inhibition,74 suggesting that multiple mutations might serve as biomarkers of worsening genetic instability or in fact cooperate functionally in varying degrees to accelerate disease progression and/or influence treatment response.
Clonality studies, including recent work interrogating MPN clones at single-cell resolution, have provided significant insights into the genetic events surrounding clonal evolution in MPNs and the order in which mutations, including MPN driver mutations, are acquired. Importantly, JAK2V617F itself can occur as a secondary event in the life of a diseased clone, lending support to the hypothesis that an earlier somatic event, such as ASXL1 or TET2 mutation, provides “fertile ground” upon which additional alterations (including JAK2V617F) are acquired (Figure 2).60,75,76 Certain mutations appear more likely to precede JAK2V617F than to follow and vice versa. Performing genomic analysis on paired MPN samples, Lundberg et al demonstrated that mutations in epigenetic modifiers TET2 and DNMT3A most commonly occur prior to the acquisition of JAK2V617F (or coexist as a separate, biclonal process) whereas mutations in IDH1/2 generally occur after JAK2V617F.60 Similarly, other studies suggest that, in contrast to de novo disease, ASXL1 mutations might occur as a late event,13,77 and a recent study incorporating combined molecular/gene-expression data at the single-cell level suggests earlier acquisition of spliceosome mutations (ie, prior to JAK2V617F).77 However, these data are complex, as Abdel-Wahab et al also previously observed that TET2 can occur after JAK2V617F in the process of leukemic transformation, suggesting a distinct role for disease alleles at different time points in the transformation process.65 Indeed, Ortmann et al observed that MPN patients who had developed a TET2 mutation prior to JAK2V617F were significantly older at time of MPN diagnosis, presented more often in chronic-phase disease (particularly ET), and exhibited a greater expansion of single-mutant hematopoietic stem cells, whereas those who had developed JAK2V617F first were more likely to be younger at diagnosis, have a PV phenotype, and demonstrate a more proliferative double-mutant progenitor population marked by excessive megakaryopoiesis and erythropoiesis.78 How mutational order might influence stem cell biology and disease progression in MPNs remains unclear, but the authors provide compelling evidence to suggest that transcriptional changes occurring as a result of the first mutation might influence the affected cell’s response to the second (or even predispose to the acquisition of it).78,79
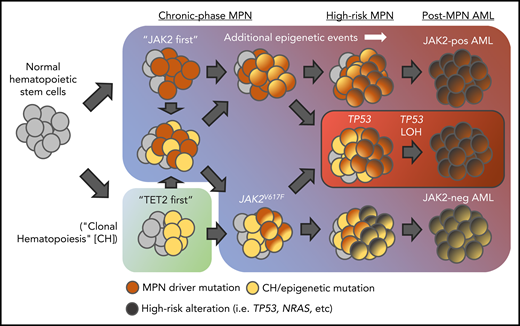
Various routes to leukemic transformation in MPNs. Mutations in epigenetic modifiers (ie, TET2, ASXL1, etc) can be acquired before or after JAK2V617F. Over time, additional somatic genetic and/or epigenetic remodeling events, under influence of various cellular-intrinsic or -extrinsic factors, promote progressive proliferative and self-renewal capacity upon the expanding cell population, ultimately leading to blast-phase transformation. Notably, in many instances, leukemic clones arise from a JAK2V617F wild-type (yellow) cell population suggesting evolution of a separate, coexisting clonal process. In the case of TP53-mutant post-MPN AML (red box), literature suggests a distinct route to leukemic transformation in which “second-hit” loss of heterozygosity (LOH) of TP53 in a preexisting JAK2V617F/TP53 heterozygous-mutant clone results in rapid clonal expansion, chromosomal instability, and blast-phase transformation.
Various routes to leukemic transformation in MPNs. Mutations in epigenetic modifiers (ie, TET2, ASXL1, etc) can be acquired before or after JAK2V617F. Over time, additional somatic genetic and/or epigenetic remodeling events, under influence of various cellular-intrinsic or -extrinsic factors, promote progressive proliferative and self-renewal capacity upon the expanding cell population, ultimately leading to blast-phase transformation. Notably, in many instances, leukemic clones arise from a JAK2V617F wild-type (yellow) cell population suggesting evolution of a separate, coexisting clonal process. In the case of TP53-mutant post-MPN AML (red box), literature suggests a distinct route to leukemic transformation in which “second-hit” loss of heterozygosity (LOH) of TP53 in a preexisting JAK2V617F/TP53 heterozygous-mutant clone results in rapid clonal expansion, chromosomal instability, and blast-phase transformation.
Despite improved understanding of the molecular heterogeneity and clonal evolution of MPNs, the specific genetic and epigenetic events governing blast transformation remain unclear. Clinical data80 as well as preclinical modeling81 of cooperating alleles with JAK2V617F would suggest that specific mutation combinations with JAK2V617F alone are insufficient to engender leukemic transformation and that additional, still unknown, genetic and/or epigenetic changes are necessary. Patients with combined JAK2V617F/IDH1/2 mutations, for example, have some of the highest rates of AML progression; however, these cells were also observed to remain chronic for years before manifesting as leukemic disease.70 This is also perhaps further evidenced by the frequent occurrence of JAK2V617F− AML arising from a JAK2V617F+ chronic-phase MPN.13,14,82,83 In these instances, the leukemic clone likely expands from a separate, coexisting JAK2V617F− diseased cell population (perhaps under selective pressure from the JAK2-mutated clone itself). Beer et al show evidence to suggest that JAK2 wild-type AMLs tend to evolve more directly from a chronic-phase ET/PV whereas JAK2-mutant AMLs are more associated with a preleukemic fibrotic stage.84 These data suggest that the antecedent disease state itself and/or other cell-extrinsic mechanisms (ie, tumor microenvironment) exert differential selection pressures to varying degrees on individual subclones to promote leukemia development.
TP53 mutations provide an example of how a singular molecular event might directly influence leukemic progression in MPNs. Mutations of TP53 have been found to coexist frequently with JAK2V617F at low, stable variant allele frequencies in patients with chronic-phase disease,13,60,85 and variant allele frequencies <5% were not found to significantly impact OS in the study follow-up period.85 These data suggest that low-abundance TP53 mutations do not necessarily influence disease progression in MPNs and that JAK2V617F and TP53 haploinsufficiency together are insufficient for blast-phase transformation. Using paired patient samples, however, Lundberg et al60 and Rampal et al13 observed a dramatic increase in mutant TP53 allele fractions following loss of the remaining wild-type TP53 allele (by chromosomal deletion or mitotic recombination) at time of leukemic transformation, supporting the notion that JAK2V617F and TP53 nullizygosity are sufficient to induce MPN blast-phase transformation (irrespective of any additional co-occurring mutations that might also be present).13,60 This was validated in vivo by expressing Jak2V617F in tp53 knockout mice. Whereas Jak2V617F/tp53 heterozygous mice (in which a tp53 wild-type allele is retained) failed to develop transformed MPN disease, Jak2V617F mice in a tp53 full knockout background develop a rapid, progressive, lethal erythroleukemia with significantly impaired OS.13,86
Together, these data highlight the complexity of clonal evolution in MPNs and the lack of our current understanding of the individual processes promoting leukemic transformation. Clonality studies confirm that linear and branched evolutionary patterns likely exist for MPN as they do for other myeloid diseases, including de novo AML. At the same time, mutational order in MPNs appears to run counter to that seen in de novo disease in that JAK-STAT–activating mutations generally occur earlier in MPNs (at least, prior to leukemic transformation) whereas signaling pathway mutations in de novo AML (eg, FLT3, KRAS, KIT, etc) are considered late events. Similarly, epigenetic mutations, including frequently mutated clonal hematopoiesis genes TET2, DNMT3A, and ASXL1, appear to be acquired earlier in de novo AML but can be acquired/expanded at time of leukemic transformation in post MPN-AML. The biological explanation for this remains unclear but likely has important pathophysiologic (and potential therapeutic) implications.
Although further work is required to ascertain the mechanisms driving clonal evolution in MPNs, our understanding of the molecular contributions to MPN disease heterogeneity is changing how we better identify patients at high risk for progression. Newer prediction scoring systems combining extended molecular profiling with traditional clinical and cytogenetic risk features, such as the Mutation-Enhanced International Prognostic System Plus (MIPSS+) v2.021 and Genetically Inspired Prognostic Scoring System (GIPSS)22 calculators, show promise in using genetic data to guide treatments. Perhaps the most exciting recent development was work by Grinfeld et al, who performed extended mutational profiling in a large cohort of MPN patients to develop a highly personalized, predictive scoring calculator tailored to the unique molecular makeup of an individual patient.20 Validation using an external cohort suggested superiority in predictive accuracy of this method in comparison with currently used International Prognostic Scoring System (IPSS) and Dynamic International Prognostic Scoring System Plus (DIPSS-Plus) models. Although prospective trials and longitudinal study will be essential to substantiating these findings, this study highlights the potential power of molecular genotyping over conventional methods alone in influencing MPN prognostication and treatment decision-making.
Current treatment options for post-MPN AML
Despite improved understanding of the molecular events involved in clonal evolution of blast-phase MPNs, advances in treatment outcomes for post-MPN AML have unfortunately languished. There are still no current standard guidelines for the management of post-MPN AML, and outcomes remain generally poor. Treatment options generally consist of noncurative low-intensity therapies, standard AML-intensive induction regimens, and allogeneic stem cell transplant (alloSCT) and are generally tailored to individual patients based on their age, functional status, and underlying comorbidities at time of leukemic progression.37,38 Patients presenting with high-risk features (ie, by DIPSS criteria) or in accelerated phase are generally warranted therapy options similar to those with transformed disease. Given the rarity of this patient population, there are no prospective trials of post-MPN AML management, and retrospective cohorts are often limited in size and by patient heterogeneity. No clinical improvements have been made for the treatment of blast-phase MPN over the last 15 years,18 underscoring the urgent need for novel therapeutic strategies and prospective clinical trials.
To date, alloSCT remains the only treatment option shown to have any meaningful prolongation in survival for post-MPN AML and is currently recommended for all fit patients for whom an adequate donor is available. This is based on several retrospective series demonstrating superior survival outcomes of alloSCT over other modalities, with 2-year OS rates ranging from 29% to 75% for alloSCT vs 0% to 15% for systemic treatments alone (Table 2).8,19,26,71,87-92 Although some analyses have suggested complete response (CR)/CR with incomplete hematologic recovery (CRi) rates following induction therapy might predict for favorable outcomes posttransplant,19,71,88,89 this observation has not necessarily held true across all studies,8,18,64 likely owing to the heterogeneity of patient cohorts evaluated. Furthermore, recent series of large patient cohorts providing extended follow-up out to 3 to 5 years suggest that long-term survival post-alloSCT remains woefully inadequate. Tefferi et al and Cahu et al, for example, each reported 3-year post-alloSCT survival rates of only 33% and 18%, respectively, which decreased further in the Tefferi study to only 10% at 5 years,18,19 highlighting the current limitations of this therapy.
Summary of key retrospective series of post-MPN AML
| Study | Year | N | Treatment | CR/CRi, % | mOS in months/OS |
|---|---|---|---|---|---|
| Supportive care regimens | |||||
| Tam et al8 | 2008 | 19 | Transfusion alone ± hydroxyurea | 0 | 1.5/5% (@ 1 y) |
| Venton et al71 | 2018 | 28 | 0 | 1.8 | |
| Mesa et al25 | 2005 | 48 | Transfusion alone ± hydroxyurea | 0 | 2.0 |
| McNamara et al61 | 2018 | 51 | 0 | 2.0/9% (@ 2 y) | |
| Passamonti et al93 | 2005 | 7 | 0 | 2.5 | |
| Low-intensity regimens/HMA therapy | |||||
| Passamonti et al93 | 2005 | 8 | Low-dose chemo: 6-thyoguanine, cytarabine | NR | 2.5 |
| — | HMA: N/A | — | — | ||
| Mesa et al25 | 2005 | 19 | Low-dose chemo: vincristine, melphalan/busulfan, cytarabine | NR | 2.9 |
| — | HMA: N/A | — | — | ||
| Kennedy et al26 | 2013 | 5 | Low-dose chemo: low-dose cytarabine, others | 6.3 | 6.6 |
| 6 | HMA: azacytidine, decitabine | ||||
| Badar et al96 | 2015 | — | Low-dose chemo: N/A | — | — |
| 21 | HMA: decitabine (± ruxolitinib or gemtuzumab) | 14 | 6.9 | ||
| Tam et al8 | 2008 | 8 | Low-dose chemo: gemtuzumab, vincristine + pred, other | 0 | 7.0 |
| 4 | HMA: azacytidine (+ valproic acid + ATRA), decitabine | ||||
| Chihara et al92 | 2016 | 52 | Low-dose chemo: low-dose cytarabine | 36 | 8.0 |
| 99 | HMA: (not defined) | 30 | 7.8 | ||
| Venton et al71 | 2018 | — | Low-dose chemo: N/A | — | — |
| 11 | HMA: azacytidine | NR | 7.9 | ||
| Thepot et al114 | 2010 | — | Low-dose chemo: N/A | — | — |
| 26 | HMA: azacytidine (7 d) | 16 | 8.0 | ||
| Mascarenhas et al87 | 2010 | — | Low-dose chemo: N/A | — | — |
| 6 | HMA: decitabine (5 d) | 50 | >9 | ||
| Andriani et al94 | 2015 | — | Low-dose chemo: N/A | — | — |
| 19 | HMA: azacytidine | 26 | 9.9/37% (@ 1 y) | ||
| Induction chemotherapy regimens | |||||
| Mesa et al25 | 2005 | 24 | Cytarabine ± anthracycline | 0 | 3.9 |
| Passamonti et al93 | 2005 | 8 | Cytarabine ± idarubicin, or fludarabine-based | 13 | 5.6 |
| Tam et al8 | 2008 | 36 | Cytarabine ± anthracycline | 46 | 6.0 |
| Noor et al27 | 2011 | 20 | Cytarabine + anthracycline | 0 | 6.0/30% (@ 1 y) |
| Chihara et al92 | 2016 | 71 | High-dose cytarabine | 39 | 7.1 |
| Venton et al71 | 2018 | 34 | NR | 8.3 | |
| Kennedy et al26 | 2013 | 13 | Cytarabine + anthracycline or NOVE-HIDAC | 47 | 9.4/15% (@ 2 y) |
| Allogeneic stem cell transplant | |||||
| Chihara et al92 | 2016 | 46 | 15.3 | ||
| Tefferi et al18 | 2018 | 24 | 17.5/32% (@ 3 y) | ||
| Mascarenhas et al87 | 2010 | 5 | >18/53% (@ 2 y) | ||
| Cahu et al19 | 2014 | 60 | NR/18% (@ 3 y) | ||
| Alchalby et al89 | 2014 | 46 | NR/33% (@ 3 y) | ||
| Cherington et al88 | 2012 | 8 | >20/75% [PFS] (@ 2 y) | ||
| Ciurea et al91 | 2010 | 14 | 31/49% (@ 2 y) | ||
| Tam et al8 | 2008 | 8 | >31/73% (@ 2 y) | ||
| Kennedy et al26 | 2013 | 17 | 47/47% (@ 2 y) |
| Study | Year | N | Treatment | CR/CRi, % | mOS in months/OS |
|---|---|---|---|---|---|
| Supportive care regimens | |||||
| Tam et al8 | 2008 | 19 | Transfusion alone ± hydroxyurea | 0 | 1.5/5% (@ 1 y) |
| Venton et al71 | 2018 | 28 | 0 | 1.8 | |
| Mesa et al25 | 2005 | 48 | Transfusion alone ± hydroxyurea | 0 | 2.0 |
| McNamara et al61 | 2018 | 51 | 0 | 2.0/9% (@ 2 y) | |
| Passamonti et al93 | 2005 | 7 | 0 | 2.5 | |
| Low-intensity regimens/HMA therapy | |||||
| Passamonti et al93 | 2005 | 8 | Low-dose chemo: 6-thyoguanine, cytarabine | NR | 2.5 |
| — | HMA: N/A | — | — | ||
| Mesa et al25 | 2005 | 19 | Low-dose chemo: vincristine, melphalan/busulfan, cytarabine | NR | 2.9 |
| — | HMA: N/A | — | — | ||
| Kennedy et al26 | 2013 | 5 | Low-dose chemo: low-dose cytarabine, others | 6.3 | 6.6 |
| 6 | HMA: azacytidine, decitabine | ||||
| Badar et al96 | 2015 | — | Low-dose chemo: N/A | — | — |
| 21 | HMA: decitabine (± ruxolitinib or gemtuzumab) | 14 | 6.9 | ||
| Tam et al8 | 2008 | 8 | Low-dose chemo: gemtuzumab, vincristine + pred, other | 0 | 7.0 |
| 4 | HMA: azacytidine (+ valproic acid + ATRA), decitabine | ||||
| Chihara et al92 | 2016 | 52 | Low-dose chemo: low-dose cytarabine | 36 | 8.0 |
| 99 | HMA: (not defined) | 30 | 7.8 | ||
| Venton et al71 | 2018 | — | Low-dose chemo: N/A | — | — |
| 11 | HMA: azacytidine | NR | 7.9 | ||
| Thepot et al114 | 2010 | — | Low-dose chemo: N/A | — | — |
| 26 | HMA: azacytidine (7 d) | 16 | 8.0 | ||
| Mascarenhas et al87 | 2010 | — | Low-dose chemo: N/A | — | — |
| 6 | HMA: decitabine (5 d) | 50 | >9 | ||
| Andriani et al94 | 2015 | — | Low-dose chemo: N/A | — | — |
| 19 | HMA: azacytidine | 26 | 9.9/37% (@ 1 y) | ||
| Induction chemotherapy regimens | |||||
| Mesa et al25 | 2005 | 24 | Cytarabine ± anthracycline | 0 | 3.9 |
| Passamonti et al93 | 2005 | 8 | Cytarabine ± idarubicin, or fludarabine-based | 13 | 5.6 |
| Tam et al8 | 2008 | 36 | Cytarabine ± anthracycline | 46 | 6.0 |
| Noor et al27 | 2011 | 20 | Cytarabine + anthracycline | 0 | 6.0/30% (@ 1 y) |
| Chihara et al92 | 2016 | 71 | High-dose cytarabine | 39 | 7.1 |
| Venton et al71 | 2018 | 34 | NR | 8.3 | |
| Kennedy et al26 | 2013 | 13 | Cytarabine + anthracycline or NOVE-HIDAC | 47 | 9.4/15% (@ 2 y) |
| Allogeneic stem cell transplant | |||||
| Chihara et al92 | 2016 | 46 | 15.3 | ||
| Tefferi et al18 | 2018 | 24 | 17.5/32% (@ 3 y) | ||
| Mascarenhas et al87 | 2010 | 5 | >18/53% (@ 2 y) | ||
| Cahu et al19 | 2014 | 60 | NR/18% (@ 3 y) | ||
| Alchalby et al89 | 2014 | 46 | NR/33% (@ 3 y) | ||
| Cherington et al88 | 2012 | 8 | >20/75% [PFS] (@ 2 y) | ||
| Ciurea et al91 | 2010 | 14 | 31/49% (@ 2 y) | ||
| Tam et al8 | 2008 | 8 | >31/73% (@ 2 y) | ||
| Kennedy et al26 | 2013 | 17 | 47/47% (@ 2 y) |
—, not applicable; @, at; ATRA, all-trans retinoic acid; chemo, chemotherapy; mOS, median overall survival; N/A, not applicable; NOVE-HIDAC, nilotinib combined with mitoxantrone, etoposide, and high-dose cytarabine; NR, not reported; PFS, progression-free survival; pred, prednisone.
For nontransplant candidates, systemic treatments, including both traditional AML-style induction regimens as well as less intensive options (ie, low-dose chemotherapy, hypomethylating agents [HMAs], and/or JAK2 inhibitors), remain viable, albeit limited, alternatives. Low-dose cytarabine appears to be demonstrably ineffective across multiple studies, with median survival rates on par to that of supportive care alone (ie, 1.5-5 months).8,25,93 Traditional AML induction regimens are only modestly better; contemporary studies demonstrate median OS durations ranging from only 3.9 months to 9.4 months,8,25-27,92-94 and relapse rates remain high. Why post-MPN leukemias respond so poorly to induction chemotherapy in comparison with de novo disease remains unclear but is likely related in part to enhanced genetic instability and/or cell-adaptive survival programs acquired over time by the diseased clone conferring chemotherapy resistance, as is often observed in other secondary/treatment-related AMLs.
HMA therapy is increasingly recognized as an alternative to induction chemotherapy for post-MPN AML treatment, particularly for unfit patients. Promoter hypermethylation of the p16ink4A locus, essential for cell-cycle regulation via the Rb-E2F–signaling pathway, has been implicated in MPN leukemic progression95 providing a rationale for HMA therapy in this setting and other myeloid malignancies. Notably, increasing evidence suggests that HMA therapy might provide clinical outcomes comparable to that of induction-style regimens but with overall improved tolerability. In 1 retrospective series of 73 post-MPN AML patients receiving azacytidine vs chemotherapy, nonsignificant differences were observed in both response rate (58.8% for induction chemotherapy vs 54.6% azacytidine) and median OS (8.3 months vs 7.9 months, respectively).71 Similar results were found in another recent study evaluating decitabine (7.6 months with induction vs 6.9 months with decitabine).96 Furthermore, rare, durable responses to HMA therapy have also been observed, extending beyond 45 months in a single patient in 1 series, again highlighting the potential benefits of HMA therapy for treatment of blast-phase MPN.87,92
Finally, individual groups have also evaluated the role of ruxolitinib for the treatment of post-MPN AML. In a phase 2 prospective trial of 38 patients with refractory acute leukemia (including 18 with post-MPN AML), 2 rounds of treatment with 25 mg or 50 mg of ruxolitinib twice daily induced a response rate of 16%, with 3 of 18 patients achieving CR/CRi and an associated improvement in spleen size.97 Response did not correlate with significant reductions in the JAK2V617F mutant allele fraction, however, suggesting pleotropic effects of JAK2 inhibitor therapy. Regardless, in a follow-up trial evaluating 3 dosing levels of ruxolitinib (50 mg, 100 mg, and 200 mg twice daily) in a similar, heavily pretreated population of secondary AML patients (including 16 of 27 with previous myelodysplastic syndrome or MPN), no major response was observed for the post-MPN AML cohort98 suggesting JAK2 inhibitor therapy alone in this setting might be insufficient to achieve any meaningful disease-modifying activity.
Emerging treatment considerations
Given the overall poor response to current treatments, there is an ongoing need for newer and smarter therapies. Novel tractable combination strategies and epigenetic-modifying therapies are currently being evaluated for the treatment or prevention of blast-phase MPN with some showing early promise.
On the basis of the efficacy of decitabine and ruxolitinib as monotherapy in accelerated-phase and blast-phase MPN, as well as preclinical evidence to suggest synergistic activity of combined JAK2 inhibitor and HMA on JAK2-mutant leukemia cell growth in vitro,13 Rampal et al recently conducted a prospective phase 1 trial of combined 5-day decitabine with escalating doses (10 mg, 15 mg, 25 mg, 50 mg) of ruxolitinib for patients with post-MPN AML.99 The combination was overall well tolerated but associated with a greater degree of hematologic toxicity at increased doses of ruxolitinib. Notably, the response rate was 57% (9 of 17 patients) with CR/CRi in 4 of 17 patients (23.5%), and a median OS of 7.9 months was observed. In addition, a reduction in blast count was seen in all patients. In light of these potentially promising findings, a new multi-institutional phase 2 study is currently under way evaluating decitabine plus ruxolitinib 10 mg twice daily.
Isocitrate dehydrogenase (IDH) inhibition represents an attractive target for the treatment of IDH-mutant post-MPN AML and/or high-risk chronic-phase disease, highlighting the potential for combined mutation-directed precision therapy for specific MPN/post-MPN AML genotypes. Gain-of-function IDH1/2 mutations are present in 15% to 25% of patients with de novo AML, and small molecule IDH inhibitors have shown significant single-agent efficacy in the relapsed/refractory setting, including in heavily pretreated patients.100-102 Given the frequent enrichment of IDH1/2 mutations at time of leukemic transformation in MPNs,13,62,68 there is a clinical rationale for the use of IDH inhibitors for the treatment of IDH1/2-mutated blast-phase/accelerated-phase disease. In addition, there is also preclinical evidence to suggest efficacy of combined JAK2 and IDH inhibition on tumor growth inhibition and suppression of abnormal 2-hydroxyglutarate production in in vivo models of JAK2V617F/IDH2 MPNs.103 As such, clinical trials for advanced IDH-mutant MPN, including post-MPL AML, are ongoing, and recent retrospective data of IDH1/2 inhibitor use in the post-MPN AML setting suggests early promise.104
Similarly, bromodomain and extraterminal domain (BET) protein inhibition offers another potential option for the treatment of advanced forms of MPN, including post-MPN AML. BET proteins are chromatin modifiers implicated in the regulation of AML cell growth, and BET inhibition has been evaluated for use in de novo AML in the relapsed/refractory setting.105 Notably, Saenz et al found that treatment of primary CD34+ blasts from post-MPN AML patients with combined BET inhibitor (JQ1) and ruxolitinib therapy resulted in enhanced cell apoptosis and growth arrest in vitro over either single-agent therapy alone.106 Furthermore, recent studies show that combined JAK2/BET inhibition can ameliorate fibrosis and prolong survival in preclinical animal models of myelofibrosis, in part through attenuation of NF-κB signaling.107 These data together provide justification for the use of combined JAK2/BET inhibition in high-risk MPN, particularly PMF, as well as for blast-phase disease, and similar trials are currently under way.
The inherently poor outcomes of post-MPN AML will likely warrant the need for additional innovative treatment strategies to more effectively eradicate post-MPN AML clones in the years to come. Bcl2 inhibitors, such as venetoclax, have shown remarkable activity in older adults with de novo AML,108,109 and several studies have identified dysregulated BclXL protein expression and apoptotic pathway signaling in MPNs,110,111 suggesting a role for Bcl2 plus or minus BclXL inhibition for the treatment of blast-phase disease. Although recent studies have identified increased programmed death ligand 1 (PDL1) expression on MPN cells with associated impairment of anti-MPN T-cell immune responses,112 the basis for immune recognition in low mutational rate myeloid malignancies such as MPN has not been demonstrated, and sufficient preclinical/clinical data demonstrating the utility of immunotherapy in blast-phase MPNs, including anti-PDL1 antibody treatment, is still lacking. Other exciting areas of active research that might provide additional insights into how to better target advanced-phase MPN clones include (1) proinflammatory cytokine signaling, fibrosis development, and clonal evolution within the context of the bone marrow microenvironment, (2) the role of reactive oxygen species, DNA repair, and modulation of p53/MDM2 pathway signaling, and (3) chromatin dysfunction and epigenetic alteration of specific transcriptional states to identify novel synthetic lethal vulnerabilities.
Conclusion
Leukemic transformation following chronic-phase MPN remains an important clinical and diagnostic dilemma. We continue to refine how to best identify those MPN patients at greatest risk for disease progression. Although alloSCT has the potential to result in long-term clinical benefit, only a minority of patients are able to receive this therapy, and relapse rates remain high, underscoring the significant clinical unmet need for these affected individuals. Novel prognostication scoring systems incorporating traditional clinical and cytogenetic risk features with molecular genotyping provide promise in helping to better stratify patients who would benefit from earlier intervention; however, further validation will be essential before these models can be widely adopted by practitioners.
Although advancements in genomic techniques have revealed the genetic complexity and heterogeneity of MPNs and expanded our understanding of the somatic alterations important for disease evolution, there remain significant gaps in our understanding of how changes at the molecular level alter the life course of the diseased cell. The enrichment of mutations in epigenetic modifiers at transformation, including TET2, ASXL1, IDH1/2, and EZH2, highlights the need to better understand how DNA methylation and chromatin dynamics influence MPN stem cell fitness and cooperate with JAK2V617F and other MPN drivers to promote clonal expansion. With the exception of JAK2V617F/TP53 mutant post-MPN AML, current preclinical animal models fail to recapitulate the detrimental effects of combined mutation on leukemic transformation in MPNs. New and innovative preclinical models of MPN and post-MPN AML are needed to more accurately model the full sequence of genetic events with temporal and spatial control. In so doing, we might uncover novel therapeutic targets better capable of improving the outcomes of our patients.
Acknowledgments
This work was supported by Memorial Sloan Kettering Cancer Center support grant P30 CA008748 from the National Institutes of Health, National Cancer Institute, and support from National Institutes of Health, National Cancer Institute grant P01 CA108671 (R.L.L.).
Authorship
Contribution: All authors contributed equally to the preparation, editing, and finalization of the manuscript text and figures.
Conflict-of-interest disclosure: A.J.D. received funding from the American Society of Clinical Oncology (ASCO) and American Association of Cancer Research (AACR). R.K.R. has received consulting fees from Incyte, Celgene, Constellation, Agios, Jazz, BeyondSpring, and Partner Therapeutics, and research funding from Incyte, Stemline and Constellation. R.L. is on the supervisory board of Qiagen; is a scientific advisor to Loxo, Imago, C4 Therapeutics, and Isoplexis, each of which includes equity interest; receives research support from and consulted for Celgene and Roche; received research support from Prelude Therapeutics; has consulted for Lilly, Janssen, Incyte, Novartis, and Gilead; and has received honoraria from Lilly and Amgen for invited lectures.
Correspondence: Ross Levine, Memorial Sloan Kettering Cancer Center, 1275 York Ave, Box 20, New York, NY 10065; e-mail: leviner@mskcc.org.
