


Abstract
Vitamin C serves as a cofactor for Fe(II) and 2-oxoglutarate–dependent dioxygenases including TET family enzymes, which catalyze the oxidation of 5-methylcytosine into 5-hydroxymethylcytosine and further oxidize methylcytosines. Loss-of-function mutations in epigenetic regulators such as TET genes are prevalent in hematopoietic malignancies. Vitamin C deficiency is frequently observed in cancer patients. In this review, we discuss the role of vitamin C and TET proteins in cancer, with a focus on hematopoietic malignancies, T regulatory cells, and other immune system cells.
Introduction
Vitamin C is an essential dietary supplement for humans.1-4 Although plants and other organisms use several pathways for synthesis of vitamin C from monosaccharides including mannose, fructose, and galactose, most vertebrates and mammals synthesize vitamin C primarily from glucose, through a pathway whose final step is catalyzed by the enzyme l-gulonolactone oxidase (GULO; Figure 1). Humans and higher primates lack a functional form of GULO, and hence vitamin C must be supplied through the diet. Vitamin C deficiency results in scurvy (scorbutus), a disease characterized by bleeding gums and poor wound healing, which was once common in sailors at sea whose diet was lacking in fresh fruits (especially citrus fruits) and vegetables. In the most recent National Health and Nutritional Examination Survey,5 fully 7% of the US population was found to be vitamin C deficient, with serum vitamin C concentrations approaching scorbutic levels (<11.4 µM). In contrast, in healthy adults ingesting adequate amounts of vitamin C a day, plasma levels of vitamin C ranged between 50 and 80 µM.1-5 There was no correlation of vitamin C deficiency with increasing age, because the highest serum vitamin C concentrations were found in children (6-11 years), followed by older individuals (>60 years), a metric that may correlate with taking supplemental vitamins.5 The implications of these estimates for clonal hematopoiesis and cancer susceptibility are considered below.
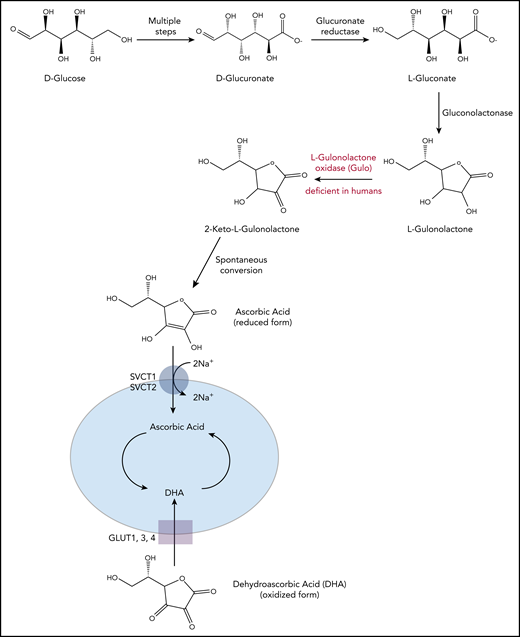
Vitamin C biosynthesis pathway and transportation in animals. Vitamin C is synthesized from d-glucose in most mammals. Through multiple enzymatic reactions, d-glucose is first oxidized to d-glucuronate, which is then reduced to l-gluconate and further into l-gulonolactone. In the final step, the enzyme GULO oxidizes l-gulonolactone to 2-keto-l-gulonolactone, which is then spontaneously converted into vitamin C (ascorbic acid). Humans, higher primates, guinea pigs, and fruit bats lack a functional form of GULO and therefore require dietary supplement. The reduced form of vitamin C, ascorbic acid, is transported into the cells through SVCT1/2 (sodium-dependent vitamin C transporters, encoded by Slc23a1 and Slc23a2), whereas its oxidized form, DHA, is transported into the cells through GLUT1/3/4 (glucose transporters). Slc23a1 is not expressed in hematopoietic cells; Slc23a2 is expressed at higher levels in HSPCs and multipotent progenitor cells compared with other, more committed hematopoietic progenitors and differentiated cell types.
Vitamin C biosynthesis pathway and transportation in animals. Vitamin C is synthesized from d-glucose in most mammals. Through multiple enzymatic reactions, d-glucose is first oxidized to d-glucuronate, which is then reduced to l-gluconate and further into l-gulonolactone. In the final step, the enzyme GULO oxidizes l-gulonolactone to 2-keto-l-gulonolactone, which is then spontaneously converted into vitamin C (ascorbic acid). Humans, higher primates, guinea pigs, and fruit bats lack a functional form of GULO and therefore require dietary supplement. The reduced form of vitamin C, ascorbic acid, is transported into the cells through SVCT1/2 (sodium-dependent vitamin C transporters, encoded by Slc23a1 and Slc23a2), whereas its oxidized form, DHA, is transported into the cells through GLUT1/3/4 (glucose transporters). Slc23a1 is not expressed in hematopoietic cells; Slc23a2 is expressed at higher levels in HSPCs and multipotent progenitor cells compared with other, more committed hematopoietic progenitors and differentiated cell types.
Vitamin C can exist in both reduced and oxidized forms.1-4 The reduced form of vitamin C is called ascorbic acid (ascorbate); this name originated from Latin with the meaning of “without scurvy.” Ascorbate enters the cells mainly through sodium-dependent vitamin C transporters (SVCTs, encoded by Slc23a1 and Slc23a2; Slc23a1 is not expressed in hematopoietic cells).4 The oxidized form of ascorbate, dehydroascorbic acid (DHA), enters cells through facilitated glucose transporters (Glut1, 3, and 4) that primarily transport glucose and are not specific for transporting DHA (Figure 1). Ascorbate is present in the plasma of healthy humans at much higher concentrations than DHA; it is taken up and concentrated by cells, so that intracellular ascorbate concentrations are in the millimolar range.1 In patients with hematologic malignancies and other cancers, however, plasma ascorbate levels tend to be exceedingly low,4,6-9 potentially because of decreased gut absorption.
Among its other biochemical functions,2,3 vitamin C facilitates the activity of Fe(II) (reduced iron, Fe2+) and 2-oxoglutarate (2OG)-dependent dioxygenases, whose catalytic activity requires the presence of reduced iron at the active site.10,11 The requirement of these enzymes for ascorbate is not strict,11 and ascorbate is rarely needed in the first cycles of catalysis in vitro12 ; rather, it appears to counter various side reactions that result in irreversible oxidation of iron, thus promoting reductive regeneration of Fe(II) at the enzymatic active site.3 The prototypical member of the Fe(II)-2OG–dependent dioxygenase family is collagen proly-4-hydroxylase10,11 ; decreased function of this enzyme because of vitamin C deficiency leads to incomplete hydroxylation of proline residues in collagen and the consequent development of scurvy.1-4 Vitamin C also potentiates the activities of several Fe(II)-2OG–dependent dioxygenases involved in gene transcription and epigenetic regulation, including histone demethylases that contain Jumonji C (JmjC) domains (members of the JHDM1, 2, and 3 families) and members of the TET family of 5-methylcytosine (5mC) oxidases.3,4,10,11 TET enzymes catalyze the sequential oxidation of 5mC in DNA to 5-hydroxymethylcytosine (5hmC)12-14 and the further oxidation products 5-formylcytosine and 5-carboxylcytosine15,16 ; these oxidized methylcytosines are well established as essential intermediates in DNA demethylation12-20 (Figure 2A).
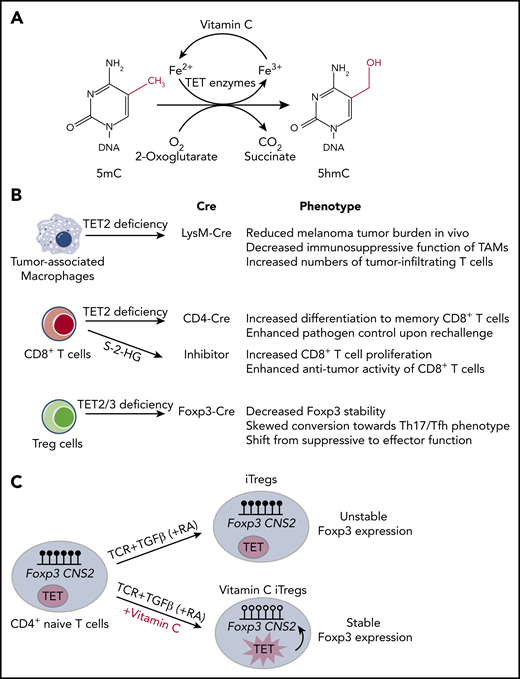
Vitamin C functions as a cofactor for TET family dioxygenases. (A) TET family dioxygenases use 2-oxoglutarate, reduced iron (Fe2+), and oxygen to oxidize 5mC into 5hmC, with CO2 and succinate as byproducts. Vitamin C functions as a cofactor for TET enzymes by facilitating the reduction of Fe3+ back to Fe2+. (B) Summary of the effects of TET deficiency in tumor-infiltrating immune cells, including tumor-associated macrophages, CD8+ T cells, and Treg cells. (C) iTregs differentiated in the presence of transforming growth factor β (TGFβ), retinoic acid (RA), and vitamin C have enhanced TET activity, which results in DNA demethylation at Foxp3 CNS2 region and increased iTreg stability.
Vitamin C functions as a cofactor for TET family dioxygenases. (A) TET family dioxygenases use 2-oxoglutarate, reduced iron (Fe2+), and oxygen to oxidize 5mC into 5hmC, with CO2 and succinate as byproducts. Vitamin C functions as a cofactor for TET enzymes by facilitating the reduction of Fe3+ back to Fe2+. (B) Summary of the effects of TET deficiency in tumor-infiltrating immune cells, including tumor-associated macrophages, CD8+ T cells, and Treg cells. (C) iTregs differentiated in the presence of transforming growth factor β (TGFβ), retinoic acid (RA), and vitamin C have enhanced TET activity, which results in DNA demethylation at Foxp3 CNS2 region and increased iTreg stability.
Several studies have shown that vitamin C promotes DNA demethylation by increasing TET-mediated oxidation of 5mC.21-24 The addition of vitamin C, but not other antioxidants, to cultures of mouse embryonic stem (ES) cells resulted in a rapid and global increase in 5hmC, which was not observed in ES cells lacking Tet1 and Tet221,23 ; the increase in 5hmC was followed by DNA demethylation at germline gene promoters.21 Addition of vitamin C to mouse fibroblasts increased 5hmC generation in a dose- and time-dependent manner.22 Similarly, addition of vitamin C to cultures of differentiating mouse and human regulatory T cells (Treg cells) maintained global 5hmC levels during differentiation without altering TET mRNA levels and promoted DNA demethylation at critical regulatory regions of Treg-specific genes; demethylation was not observed in mouse Treg cells lacking Tet2 and Tet3, indicating that vitamin C acted through TET proteins to achieve enhanced demethylation.24 Vitamin C interacted directly with the catalytic domain of Tet2, as shown by the ability of vitamin C to quench the intrinsic fluorescence of recombinant Tet2 catalytic domain.23
In this article, we discuss the potential roles of vitamin C and TET proteins in cancer, with a focus on hematopoietic malignancies. For related reviews please see references 3,4,19 , and 20 . In the context of immune responses to cancer, we also briefly discuss the importance of TET enzymes and vitamin C in regulating immune function.
TET enzymes and vitamin C in leukemogenesis
Loss-of-function mutations in TET genes, especially TET2, are prevalent in hematopoietic malignancies.25-28 Two studies have illustrated the connections between vitamin C and TET function in leukemia progression.29,30 The first study29 reported that expression of the vitamin C transporter gene Slc23a2, as well as the levels of vitamin C itself, were high in hematopoietic stem/precursor cells (HSPCs) and multipotent progenitor cells (MPPs) compared with other more committed hematopoietic progenitors and differentiated cell types. Like Tet2-deficient mice,31 vitamin C-depleted Gulo−/− mice had increased HSPC frequencies, higher levels of lineage reconstitution capacity in competitive transplantation assays, and reduced levels of 5hmC.29 In humans, TET2 mutations cooperate with FLT3ITD mutations to induce acute myeloid leukemia (AML). Strikingly, vitamin C depletion accelerated the development of AML from Flt3ITDTet2+/− and Flt3ITDTet2−/− cells, and this phenomenon was reversed by dietary repletion of vitamin C.29 The second study30 used a short hairpin RNA transgenic mouse to reversibly knock down and restore endogenous Tet2 expression. Loss of Tet2 resulted in an aberrant increase in self-renewal of hematopoietic stem/precursor cells (HSPCs), associated with increased DNA methylation, aberrant differentiation toward the myeloid lineage, and increased cell death. Restoration of Tet2 reversed these phenomena, and treatment with vitamin C pharmacologically mimicked the effects of Tet2 restoration in HSPCs and blocked the progression of myeloid disease. The effects of vitamin C were TET dependent because they disappeared when both Tet2 and Tet3 were absent. In human AML cells, vitamin C increased TET activity and induced DNA demethylation; gene set enrichment analysis showed that vitamin C-induced genes were enriched in genes upregulated on Tet2 restoration. Furthermore, vitamin C treatment increased the sensitivity to polyADP ribose polymerase (PARP) inhibition in a panel of human myeloid leukemia cells, leading the authors to conclude that vitamin C treatment, alone or combined with PARP inhibitors, could be novel therapeutic strategies for patients with hematologic cancers.30
In light of these findings, it would be interesting to examine the role of vitamin C deficiency in the development of clonal hematopoiesis (CH).32-35 The predominant mutations in CH involve loss-of-function mutations in DNMT3A, TET2, and ASXL1, the regulatory component of the ASXL1-BAP1 histone H2A deubquitinase complex.36 Vitamin C deficiency in Gulo-deficient mice is associated with increased numbers of HSPCs,29 but whether vitamin C deficiency contributes to CH in humans by decreasing TET activity in HSPCs has not been formally tested. Recent findings indicate an unexpected link between CH and cardiovascular disease,37 and experiments in mouse models of hypercholesteremia suggest that expanded myeloid cells in Tet2-deficient mice are the source of high levels of inflammatory mediators (interleukin-1β, interleukin-6), which are almost invariably observed in the blood of persons with CH.38,39 It would be useful to investigate whether certain cases of CH, especially those without the 2 major CH-associated mutations (DNMT3A and TET2), are linked to low plasma levels of vitamin C. Indeed, in the European Prospective Investigation into Cancer (EPIC)-Norfolk study involving almost 20 000 individuals, low plasma vitamin C levels were associated with an overall increased risk of mortality from all causes, including cardiovascular disease, ischemic heart disease, and cancer.40 This finding may stimulate future clinical trials of high-dose vitamin C supplementation, whether oral or intravenous (see below), in individuals with CH, with the hope of diminishing the risks of malignant progression and cardiovascular disease.
Vitamin C deficiency in cancer and the debate over supplementation with vitamin C
Cancer patients are significantly depleted of plasma vitamin C compared with healthy individuals,4,6-9 but the jury is still out on whether vitamin C deficiency is directly responsible for, or merely correlates with, an increased risk of malignancy. Because TET deficiencies, TET loss-of-function mutations, and TET loss-of-function caused by hypoxia and various regulatory and metabolic derangements are associated with many types of cancers,4,25-27,31,41-45 dietary supplementation with vitamin C might in fact be beneficial in preventing cancers with TET involvement in the long term. However, the field was mired in controversy until quite recently.2,46-51 In an early nonrandomized trial, Cameron and Pauling46 treated 100 terminal cancer patients with intravenously administered high-dose vitamin C at 10 g/day for about 10 days, with treatment continued orally thereafter; the authors concluded that high-dose vitamin C had beneficial effects on the survival time of the cancer patients. Shortly afterward, 2 randomized double-blinded clinical trials were conducted at the Mayo Clinic, in which patients with advanced cancer were given placebo or high-dose vitamin C at 10 g/day orally; these studies showed no beneficial effects with high-dose vitamin C therapy.47,48 The discrepancies were later explained by pharmacokinetic studies in healthy volunteers, showing that plasma vitamin C concentrations differed markedly depending on whether the route of administration was oral or intravenous.2,49 When vitamin C was orally administered, plasma concentrations were tightly controlled: the maximum oral dose tested, 3 g every 4 hours, yielded plasma levels in the submillimolar range (∼200 µM), whereas intravenous administration of 3 g of vitamin C yielded plasma concentrations that were 30- to 70-fold higher, in the millimolar to tens of millimolar range.2,49-51 In all cases, the high doses were well tolerated, without any safety issues. These findings suggested that the high plasma concentrations attained in the Cameron and Pauling study46 might indeed have had antitumor effects that were not observed in the follow-up trials in which only oral dosing was used.47,48 In fact, several groups have now advocated reopening the possibility of supplementation with high-dose vitamin C for the clinical treatment of cancer.1,2,4,49-51
It is still unclear how vitamin C exerts its effects on cancer cells. At physiologic levels, vitamin C is an antioxidant, but at high pharmacologic concentrations, it may promote oxidative stress and subsequent cell death. In cell culture studies, vitamin C selectively killed cancer cells but not normal cells by promoting hydrogen peroxide formation.52 Moreover, human colorectal cancer cells carrying KRAS or BRAF mutations were selectively killed when cultured with high dose vitamin C, because of accumulation of reactive oxygen species that inactivated glyceraldehyde-3-phosphate dehydrogenase (GAPDH) by glutathionylation of the active site cysteine and depletion of its substrate NAD+(the oxidized form of NAD, nicotinamide adenine dinucleotide), leading to metabolic crisis with decreased glycolysis and adenosine triphosphate and death of cancer cells, which rely more on glycolytic metabolism than their active counterparts.53 This study also proposed that the active agent for vitamin C toxicity was actually DHA, which entered cancer cells as a result of their high expression of GLUT1.53 A later study on non–small-cell lung cancer (NSCLC) and glioblastoma multiforme (GBM) cell lines suggested that the selective toxicity of high-dose ascorbate in these cell lines over normal cells was caused by vitamin C itself, because it was not reduced by competitive inhibition of GLUT1 and other glucose transporters with 2-OG.54 Rather, the toxicity was attributed to increases in intracellular redox-active labile iron pools, mediated by perturbations in cancer cell oxidative metabolism and increased steady-state levels of reactive oxygen metabolites including superoxide and hydrogen peroxide; pool sizes could be decreased and survival could be improved by knockdown of the transferrin receptor, which showed increased expression in cancer compared with normal cell lines. High-dose ascorbate combined with standard radio-chemotherapy led to the longest survival in mouse models of NSCLC and GBM, which was further demonstrated in phase 1 and phase 2 clinical trials of patients with GBM and advanced NSCLC.54 It is as likely, however, that vitamin C suppresses oncogenesis by potentiating the activity of Fe(II)-2OG–dependent dioxygenases, including TET DNA methylcytosine oxidases and JmjC domain-containing histone demethylases, as described above.21-24
Therapies that target epigenetic regulators, particularly the DNA methyltransferase inhibitors 5-azacytidine and decitabine (also known as hypomethylating agents [HMAs]), have been used successfully to treat patients with myelodysplastic syndrome (MDS), a condition that often progresses to myeloid malignancy.9,55 This clinical success story has prompted the question of whether vitamin C supplementation could also be used to improve antitumor responses in patients with frank hematopoietic and other malignancies. Vitamin C at physiologic levels clearly synergized with 5-azacytidine to decrease proliferation and increase apoptosis of several cancer cell lines in culture. The cells showed a striking increase in TET activity as judged by increased levels of 5hmC and upregulated the expression of endogenous retroviruses and concomitantly the expression of interferon-stimulated genes.9 Assuming that these features are maintained in vivo, increased TET activity and enhanced immune responses after treatment with combined 5-azacytidine and vitamin C might yield improved outcomes in MDS and other neoplastic diseases.
There have been several efforts to determine whether specific mutations in MDS could be predictive markers for responses to 5-azacytidine and decitabine. An early study with only a few patients showed no significant association of TET2 mutational status with response to 5-azacytidine: 46% (5 of 11) TET2-mutated patients but only 24% (5 of 21) TET2 wildtype patients responded to epigenetic treatment, but because of the relatively small sample size, this difference did not reach statistical significance (P = .2).56 In a parallel study published in the same volume, the authors sequenced TET2 in 86 MDS and AML patients treated with 5-azacytidine and found a significantly higher response rate in patients with TET2 mutations (82%) compared with patients without TET2 mutations (45%); however, this failed to translate into a beneficial response duration or overall survival.57 A later study of 213 MDS patients screened for mutations in 40 candidate MDS-associated genes and suggested that patients with TET2 mutations showed the highest responses to 5-azacytidine and decitabine, particularly if they lacked mutations in ASXL1.55 Two years later, yet another paper58 examined the mutational status of 26 candidate MDS genes in 107 patients with MDS and found that no single mutation or combination of mutations was significantly associated with responses to HMAs. In short, there is still no widely accepted consensus on whether individual mutations might be used as biomarkers to predict responses to HMA treatment in MDS or AML.
Hypomethylating agents cause reactivation of endogenous retroviruses and enhanced interferon responses,59,60 which in turn lead to increased production of proinflammatory cytokines and enhanced immune responses that are thought to be protective in MDS and AML. However, there are 2 caveats to keep in mind. Reactivation of endogenous retroviruses has been associated with increased mutational burden in cancer and autoimmune disease.61,62 Second, microbial signals elicit increased production of some of the same proinflammatory cytokines, which promote preleukemic expansion and eventually the malignant transformation of TET2-mutant myeloid-lineage cells in mice.63 The detailed underlying mechanisms are clearly different in the 2 scenarios (response to microbial signals vs responses to hypomethylating agents) and the preleukemic myeloproliferation observed in TET2-mutant mice reflects initiation of myeloid malignancies rather than slower disease progression in response to HMAs. Further research is needed to establish whether HMA treatment might have long-term deleterious effects.
TET enzymes and vitamin C in immune function
The recent heightened interest in cancer immunotherapy has brought up the question of whether vitamin C supplementation could be used to enhance antitumor activity by potentiating immune responses in cancer patients. Although TET deficiency promotes oncogenesis in cancer cells, TET deficiency in numerous tumor-infiltrating immune cells has the opposite effect of directly or indirectly opposing tumor growth (Figure 2B). In CD8+ T cells, TET deficiency has cell-intrinsic effects: after transfer into recipient mice, Tet2-deficient CD8+ T cells,64 and CD8+ T cells treated briefly with the enantiomer S-2-HG (aka L-2HG), a potent inhibitor of Fe(II)- and 2OG-dependent dioxygenases,65 showed increased proliferation and an enhanced ability to combat infection64 and slow tumor growth65 compared with wild-type CD8+ T cells. This feature was even more striking in a patient with B cell chronic lymphocytic leukemia, who achieved complete remission after treatment with CD8+ T cells bearing a chimeric antigen receptor (CAR) against CD19.66 This patient bore a hypomorphic mutation in 1 TET2 allele; serendipitously, in the most highly expanded CAR T-cell clone, the CAR lentivirus had become integrated into the other TET2 allele. Presumably, the resulting profound decrease in TET function in this clone led to its massive expansion and superior antitumor efficacy.66 In contrast, the effects of TET deficiency in tumor-associated macrophages and myeloid-derived suppressor cells are not cell intrinsic: these myeloid cell types normally exert potent immunosuppressive effects on tumor-infiltrating T cells,67,68 but in Tet2fl/fl LysM-Cre mice, in which Tet2 deficiency is confined to myeloid cells, the intratumoral Tet2-deficient tumor-associated macrophages were considerably less immunosuppressive, thus promoting CD8+ T-effector function and inhibiting tumor growth.69
Several recent studies in mouse models have shown that high-dose vitamin C is a promising agent to potentiate the antitumor effects of immune checkpoint therapy. High-dose vitamin C increased infiltration of CD4+ and CD8+ T cells and macrophages into the tumor microenvironment; increased production of granzyme B by CD8+ T cells and natural killer cells and production of interleukin-12 by macrophages; decreased tumor growth in a T cell–dependent manner; and synergized with immune checkpoint therapy (anti-PD1 with or without anti-CTLA4) in several cancer types.70,71 Vitamin C acted on immune cells in these models, because its administration did not affect tumor growth (including growth of the B16 melanoma) in immunocompromised mice.70 However, vitamin C was also shown to enhance Tet2 catalytic activity in B16-OVA melanoma cells, markedly improving chemokine and PD-L1 expression in response to interferon-γ in a mechanism involving Tet2 recruitment to promoters through interaction with nuclear STAT1. This increase in chemokine and PD-L1 expression was associated with increased numbers of tumor-infiltrating lymphocytes and improved antitumor immunity, as well as enhanced efficacy of anti-PD-L1 immune therapy.72
Treg cells are a distinct lineage of CD4+ T cells needed to maintain immune homeostasis and prevent autoimmune diseases; they also counter the functions of several immune cell types in the tumor microenvironment.73 The effects of TET proteins and vitamin C on Treg cells have been extensively studied at the molecular level (Figure 2C). Profound TET deficiency caused by double deletion of either Tet1 and Tet2 or Tet2 and Tet3 genes with CD4Cre resulted in increased DNA methylation at 2 intronic enhancers, CNS1 and CNS2 (conserved noncoding sequences 1 and 2)24,74 ; in both humans and mice, the methylation status of CNS2 controls the stability of Foxp3 expression.75,76 Mice with Tet2 and Tet3 deficiency in T cells (using CD4Cre) displayed normal frequencies of Foxp3+ cells in the thymus but considerably reduced frequencies in the periphery, with a concomitant decrease in Treg stability and function.24 Deletion of Tet2 and Tet3 genes at a later developmental stage using Foxp3-Cre resulted in the appearance of “ex-Treg” cells that acquired effector function.77,78 By augmenting TET activity, vitamin C promoted the optimal differentiation and function of induced regulatory T cells (iTregs) generated by stimulation of naïve CD4+ T cells with antigen, transforming growth factor β and retinoic acid in vitro, resulting in DNA demethylation at the Foxp3 CNS2 enhancer and increased Foxp3 stability.24,79 Thus, from a therapeutic perspective, enhancement of TET activity using vitamin C could be used to stabilize iTreg cells generated in vitro and endogenous Treg cells expanded in vitro with interleukin-280 to counter autoimmunity,81 allograft rejection,82 and graft-versus-host disease.83
Conclusions
TET proteins and vitamin C exert a major influence in vivo on cancer cell themselves and anticancer immune responses. The finding that low plasma vitamin C levels are associated with an increased risk of mortality from both hematopoietic malignancies and cardiovascular disease should prompt an increased investigation into the role of vitamin C deficiency in clonal hematopoiesis, a premalignant condition whose links to both these conditions have become increasingly clear. It would also be worthwhile to revisit the potential beneficial effects of high-dose vitamin C therapy in the clinical management of cancer, adoptive cell therapies for cancer, and autoimmune disease.
Acknowledgments
This work was supported by National Institutes of Health, National Institute of Allergy and Infectious Diseases grant R01 AI128589 and National Institutes of Health, National Cancer Institute grant R35 CA210043 (A.R.).
Authorship
Contribution: X.Y. and A.R. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Anjana Rao, La Jolla Institute for Immunology, 9420 Athena Circle, La Jolla, CA 92037; e-mail: arao@lji.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal