

In this issue of Blood, 1 seek to identify mechanisms by which ALK+ anaplastic large cell lymphoma (ALCL) cells develop resistance to the ALK inhibitor crizotinib. Their findings implicate a novel and potentially targetable interleukin-10 (IL-10) receptor-dependent autocrine loop that bypasses ALK signaling.
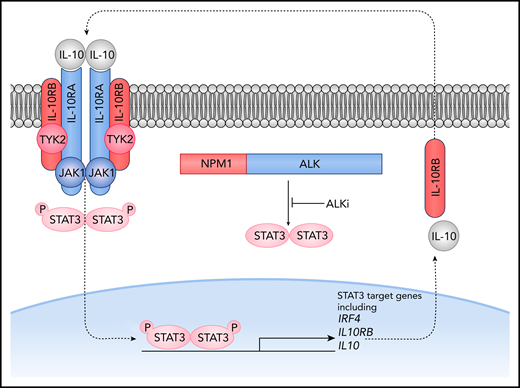
Overexpression of IL10RA leads to crizotinib resistance in ALK+ ALCL cells through an autocrine loop that bypasses the NPM-ALK fusion protein. IL-10 signaling leads to activation of STAT3, which in turn transcriptionally regulates IL10 and IL10RB and promotes cell survival despite inhibition of ALK tyrosine kinase activity. P, phosphorylation. See Figure 5H in the article by Prokoph et al that begins on page 1657.
Overexpression of IL10RA leads to crizotinib resistance in ALK+ ALCL cells through an autocrine loop that bypasses the NPM-ALK fusion protein. IL-10 signaling leads to activation of STAT3, which in turn transcriptionally regulates IL10 and IL10RB and promotes cell survival despite inhibition of ALK tyrosine kinase activity. P, phosphorylation. See Figure 5H in the article by Prokoph et al that begins on page 1657.
ALK+ ALCL is the most common T-cell non-Hodgkin lymphoma in children, the biology of which is driven predominantly by chromosomal rearrangements fusing the ALK tyrosine kinase gene to various partners, predominantly NPM1.2 The resultant fusion protein, NPM-ALK, activates numerous cellular processes, including activation of the STAT3 transcription factor. Pediatric clinical trials have honed frontline combination chemotherapy regimens for ALK+ ALCL, but toxicity and a persistent subset of patients with poor outcomes remain significant challenges. NPM-ALK also can be targeted directly. Crizotinib is an ALK/MET/ROS1 inhibitor with demonstrated clinical efficacy in pediatric ALK+ ALCL; however, crizotinib resistance develops in some patients.3 In a subset of these cases, resistance develops through acquisition of ALK mutations, which can be detected by sequencing and potentially targeted by newer-generation ALK inhibitors, such as alectinib, ceritinib, brigatinib, and lorlatinib.4 However, non-ALK signaling pathways, such as the IGF-1R pathway, also may contribute to crizotinib resistance.5 The role of these alternative signaling pathways in crizotinib resistance remains poorly understood.
To characterize mechanisms underlying crizotinib resistance in ALK+ ALCL, Prokoph et al performed a genome-wide CRISPR activation screen in 3 ALK+ ALCL cell lines and identified several candidate genes, including STAT3, RORC, MYC, IRF4, and IL10RA. The authors then evaluated these candidates by overexpressing them individually and by using a CRISPR knockout screen. The top gene candidate was IL10RA, which was overexpressed in 30% of crizotinib-resistant cell lines and was more highly expressed in a crizotinib-resistant patient sample without ALK mutations than in an ALK-mutated crizotinib-resistant patient sample or in samples from patients who relapsed on standard chemotherapy. The IL-10 receptor complex consists of a tetramer of IL10RA and IL10RB that interacts with JAK1 and TYK2 to activate STAT3 (see figure). Prokoph et al mechanistically demonstrated that the IL-10 signaling pathway modulated sensitivity of ALK+ ALCL cells to ALK inhibition via NPM-ALK–independent activation of STAT3. They further showed that STAT3 binds to the promoters of the IL10, IL10RA, and IL10RB genes, supporting the existence of a positive feedback loop that bypasses NPM-ALK. Although IL-10 signaling is common in ALK+ ALCL,6,7 only a subset of patients develops crizotinib resistance, suggesting that only particularly strong IL-10 signaling is able to maintain STAT3 activity sufficient for cell survival when NPM-ALK is inhibited by crizotinib. Notably, the existence of ALK-independent mechanisms of STAT3 activation also has been demonstrated in ALK-negative ALCL,8 although STAT3-negative ALCLs also exist.9
The findings of Prokoph et al characterize a novel and potentially targetable mechanism of crizotinib resistance in ALK+ ALCL cells. For example, JAK inhibitors, TYK2 inhibitors, and/or direct STAT3 inhibitors might contribute to regimens that treat or prevent some cases of ALK inhibitor resistance. Combining ALK inhibitors with chemotherapy also might lessen the incidence of resistance. It remains unclear, however, what proportion of clinical cases of crizotinib resistance involve the IL-10 autocrine loop demonstrated here, or what level of IL10RA expression and/or other conditions are needed in vivo for induction of this mechanism. In these studies, IL10RA was artificially overexpressed in vitro with or without IL-10 supplementation; further clinical studies will be required to assess the degree to which IL10RA overexpression by tumors represents a predominant and recurrent mechanism underlying resistance to crizotinib and perhaps other ALK inhibitors. The full diversity of ALK-independent escape mechanisms also remains unknown; survival pathways other than IL-10 signaling may exist and could relate to other gene candidates identified by the authors’ screening approach.
In summary, Prokoph et al report an elegant approach to characterize crizotinib resistance mechanisms using in vitro screening in a relatively uncommon pediatric tumor with significant barriers to obtaining large numbers of clinical samples. Ongoing characterization of the spectrum of such mechanisms could lead to new salvage regimens as well as biomarker assays that help individualize management according to the mechanism of resistance. As the authors discuss, the precise mechanism driving IL10RA overexpression as a resistance mechanism requires further study. More broadly, the factors underlying the diversity of resistance mechanisms that develop among patients with the same disease receiving the same drug remain unclear. Understanding why different tumors acquire different mechanisms of resistance, while still others retain chemosensitivity, will facilitate development of more effective tyrosine kinase inhibitors, frontline combination therapies, and personalized strategies to predict and possibly prevent resistance.
Conflict-of-interest statement: A.L.F. receives research funding from Seattle Genetics and is an inventor of technology related to ALCL for which Mayo Clinic holds unlicensed patents. G.H. declares no competing financial interests.
