

Key Points
CRISPR-modified CD38KO ex vivo expanded primary NK cells show enhanced DARA-mediated cytotoxic effect against MM.
CD38KO NK cells have increased oxidative metabolism.

Abstract
Multiple myeloma (MM) is a plasma cell neoplasm that commonly expresses CD38. Daratumumab (DARA), a human monoclonal antibody targeting CD38, has significantly improved the outcome of patients with relapsed or refractory MM, but the response is transient in most cases. Putative mechanisms of suboptimal efficacy of DARA include downregulation of CD38 expression and overexpression of complement inhibitory proteins on MM target cells as well as DARA-induced depletion of CD38high natural killer (NK) cells resulting in crippled antibody-dependent cellular cytotoxicity (ADCC). Here, we tested whether maintaining NK cell function during DARA therapy could maximize DARA-mediated ADCC against MM cells and deepen the response. We used the CRISPR/Cas9 system to delete CD38 (CD38KO) in ex vivo expanded peripheral blood NK cells. These CD38KO NK cells were completely resistant to DARA-induced fratricide, showed superior persistence in immune-deficient mice pretreated with DARA, and enhanced ADCC activity against CD38-expressing MM cell lines and primary MM cells. In addition, transcriptomic and cellular metabolic analysis demonstrated that CD38KO NK cells have unique metabolic reprogramming with higher mitochondrial respiratory capacity. Finally, we evaluated the impact of exposure to all-trans retinoic acid (ATRA) on wild-type NK and CD38KO NK cell function and highlighted potential benefits and drawbacks of combining ATRA with DARA in patients with MM. Taken together, these findings provide proof of concept that adoptive immunotherapy using ex vivo expanded CD38KO NK cells has the potential to boost DARA activity in MM.
Introduction
Multiple myeloma (MM) is characterized by clonal accumulation of malignant plasma cells in bone marrow (BM).1 Although the introduction of autologous stem cell transplantation and novel agents such as proteasome inhibitors (bortezomib and carfilzomib) as well as immunomodulatory drugs (IMiDs; lenalidomide and pomalidomide) have significantly improved survival in patients with MM, virtually all patients relapse and then suffer from poor prognosis with median overall survival of only 13 months.2,3
Most recently, monoclonal antibodies targeting CD38, daratumumab (DARA) and isatuximab, have made a significant impact on the management of patients with MM.4-6 DARA has been approved by the US Food and Drug Administration for newly diagnosed as well as relapsed/refractory (R/R) patients with MM.7-9 DARA kills target cells through several mechanisms: complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), apoptosis induced by cross-linking of CD38 on the target cells, and immunomodulatory effects via elimination of CD38+ immunosuppressive cells.10,11 Although all of these actions are involved in antitumor activity, it remains unclear which mechanism plays a major role in the clinical responses seen in patients with MM.
Despite the well-established clinical benefits of DARA, the majority of patients eventually experience disease relapse and continue to succumb to MM.12 Current research and clinical efforts are underway to unveil mechanisms of resistance to DARA and develop combination therapies to deepen or boost the response. Clinical evidence suggests that IMiDs synergize with DARA and result in better disease control.13-15 This may in part be the result of activation of natural killer (NK) cells that mediate DARA-mediated ADCC10,16,17 as well as IMiD-induced CD38 upregulation on MM cells via cereblon-mediated degradation of Ikaros and Aiolos.18
Additional evidence suggests that CD38 expression levels on target cells correlates with sensitivity to DARA. MM cells with higher CD38 expression levels are preferentially killed by DARA, whereas residual MM cells display lower CD38 expression levels during treatment with DARA.19,20 Because transcription of CD38 is directly controlled by retinoic acid via retinoic acid–responsive elements present in intron I of the CD38 gene,21 all-trans retinoic acid (ATRA) upregulates CD38 expression on a variety of hematopoietic cells including MM cells.22,23 In addition, ATRA downregulates expression of complement inhibitory proteins (CD55 and CD59) and synergizes with DARA to kill target MM cells.23 This strategy is currently being tested in a clinical trial that combines ATRA with DARA for patients with MM (ClinicalTrials.gov identifier: NCT02751255).
Another putative mechanism of suboptimal response to DARA is rapid depletion of NK cells in patients after treatment with DARA,24 because NK cells also express relatively high levels of CD38.11 This decrease in circulating NK cells persists for 3 to 6 months after discontinuation of treatment, resulting in inefficient ADCC against MM cells. Adoptive transfer of NK cells may be a strategy to overcome this mechanism. In a preclinical model, supplementation of ex vivo expanded NK cells resulted in a significant albeit modest improvement of DARA in controlling disease burden,25 probably because these NK cells are also subject to DARA-mediated elimination. An approach to overcome DARA-mediated elimination is to delete CD38 in NK (CD38KO NK) cells. Although gene editing of NK cells has been challenging because of their DNA-sensing mechanisms and associated apoptosis,26 we and others have reported efficient gene deletion in primary NK cells using a DNA-free method with Cas9 ribonucleoprotein complexes (Cas9/RNP).27,28
Here, we explore the biological consequences of CD38 deletion in NK cells with regard to DARA immunotherapy by assessing conjugation and fratricide in vitro and in vivo and ADCC against MM cells. We explored the role of CD38 as an ectoenzyme that regulates nicotinamide adenine dinucleotide (NAD+) levels29-31 in NK cell metabolism and transcription.
Materials and methods
NK cell purification and expansion
Peripheral blood (PB)–NK cells were isolated from healthy donors as previously described.32 Purified NK cells (CD3–/CD56+) were stimulated using irradiated membrane-bound interleukin-21 (IL-21; mbIL21)/4-1BBL-expressing K562 (CSTX002) at a ratio of 2:1 and grown for 7 days in AIM-V/ICSR expansion medium (CTS™AIMV™ SFM/CTS™ Immune Cell SR, Thermo Fisher Scientific) and 50 IU of human recombinant IL-2 (rIL-2) (Novartis). CD3+, CD19+, and CD33+ cells were not detected after stimulation (supplemental Figure 1A, available on the Blood Web site). Before electroporation on day 6 of expansion, half the medium was changed.27,32
MM cells
MM cell lines H929, MM.1S, U266, and RPMI 8226 were purchased from the American Type Culture Collection. OPM-2 and KMS-11 cell lines were obtained from German Collection of Microorganisms and Cell Cultures. Primary MM cells were collected from newly diagnosed or relapsed MM patients under an institutional review board–approved protocol and written consent at Johns Hopkins University. Patient information is provided in supplemental Table 1. CD138+ MM cell purification and cell culture were previously published and are detailed in supplemental Methods.33
Mice
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice were purchased from The Jackson Laboratory and were maintained under specific pathogen-free conditions. Six- to 10-week-old male NSG mice were used for experiments in accordance with our animal protocol approved by the Animal Research Committee at Johns Hopkins University.
Immunophenotyping
NK cells and MM cells were stained with fluorophore-conjugated antibodies and analyzed by flow cytometry using an LSR II flow cytometer (Becton Dickinson Biosciences) and FlowJo software (Tree Star Inc, Ashland, OR). The list of antibodies is provided in supplemental Table 2.
Generation of CRISPR-modified cells
Identifying off-target effects of CD38-targeted Cas9/RNP
Whole-genome sequencing (WGS) was performed as described in supplemental Methods and used to identify the off-target effects of Cas9/RNP targeted to CD38. Next-generation sequencing data were processed through Churchill,36 in which reads were aligned using BWA MEM (v0.7.15) to the GRCh37 reference genome. Variants were called using the Mutect2 tool of the Genome Analysis Toolkit (GATK v4.0.5.1, Broad Institute) and annotated using SnpEff (v4.3).37 Knockout-exclusive single nucleotide polymorphisms and insertion-deletion mutations (indels) were detected when compared with wild-type (WT) NK cells. Because repair of the DNA breaks generated by Cas9/RNP will vary between cells and be close to the region of guide RNA homology, clustered events were not excluded as is typically done for somatic genomic analysis. Therefore, we applied the Mutect2 filters and included those occurring at any frequency in the CD38KO cells but not present in WT NK cells, and included only those that passed all the applied filters or clustered events, nonsynonymous mutations, and in coding regions.
NK function assays
NK cell conjugation assay was performed as previously published37 and as detailed in supplemental Methods. To quantify fratricide, CD38WT and CD38KO NK cells were each treated with 10 μg/mL of DARA or solvent control for 4 or 24 hours, then stained with PO-PRO-1 dye (Invitrogen, Eugene, OR) and 7-aminoactinomycin D (Invitrogen, Eugene, OR).25 Flow-based killing assays were performed as detailed in supplemental Methods. In brief, CD38WT or CD38KO NK cells were cocultured with carboxyfluorescein succinimidyl ester–labeled target MM cells for 4 or 24 hours in the presence of 10 μg/mL of DARA or solvent as control.38
Adoptive transfer of human NK cells into NSG mice
Ex vivo expanded CD38WT and CD38KO NK cells from the same individual donor were thawed and re-stimulated with irradiated CSTX002 for 1 week. Then 107 NK cells from each group were suspended in Hank’s balanced salt solution (Gibco, Grand Island, NY) and infused through the tail vein of NSG mice that were pre-treated intraperitoneally with DARA (8 mg/kg) or solvent control on the same day. Mice were supplied with rIL-2 (50 000 IU) intraperitoneally every other day.38 PB (after 7 days), and spleen and BM (after 9 days) were collected and analyzed for the persistence of NK cells in each mouse. Absolute numbers of human NK cells in spleen and BM from 2 femurs were also calculated.
RNA-seq and IPA
RNA sequencing (RNA-seq) was performed as described in supplemental Methods. Differentially expressed genes (DEGs) were identified as those in which a paired two-sided Student t test of gene expression levels between CD38WT and CD38KO NK cells yielded a P value of < .05. Adjusting the P value cutoff for DEGs to .01 or .1 or adjusting the minimum gene expression cutoff to 5 fragments per million does not qualitatively affect conclusions. The mean fold changes of each gene across CD38WT and CD38KO NK cells are approximately equal to the fold changes of the means in the reported pathways, so inter-individual effects (CD38WT and CD38KO NK cells from the same donors) can be considered negligible for these conclusions. All default settings for a core analysis in ingenuity pathway analysis (IPA) were implemented. We were not able to study the transcriptomic profile of CD38WT and CD38KO NK cells in the presence of DARA because the CD38WT NK cells are killed by DARA-induced fratricide.
Metabolic assays
To measure the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), we used the Agilent Extracellular Flux Assay Kit (Agilent Technologies) on the Seahorse XFe24 analyzer (Agilent Technologies). Expanded CD38WT and CD38KO NK cells were incubated in XF RPMI for 1 to 2 hours before the measurements at 37°C in a non-CO2 incubator. The medium was supplemented with 10 μM glucose and 2 μM L-glutamine with no phenol red at a pH of 7.35 to 7.4. OCR, a measure of oxidative phosphorylation (OXPHOS), and ECAR were analyzed under the basal condition followed by the addition of 1 μM oligomycin, 1.5 μM carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), and 0.5 μM rotenone and antimycin A to the cell culture.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8 (GraphPad Software, Inc). Student t test was used to compare 2 independent groups. Three or more groups were compared with one-way analysis of variance test followed by Tukey’s multiple comparisons test. P < .05 was indicated statistical significance.
Results
Efficient gene targeting using Cas9/RNP in NK cells
By using the Cas9/RNP method, we successfully generated CD38KO NK cells from ex vivo expanded PB-NK cells from healthy donors (Figure 1A). Flow cytometric analysis revealed that the CD38 knockout efficiency was 81.9% ± 6.9% (n = 5; mean ± standard deviation; Figure 1B). Using magnetic separation, NK cells were purified to more than 99% CD38KO to be used for further experiments (Figure 1C). CD38WT and CD38KO NK cells showed similar expansion rates, and the purity of CD38KO NK cells was preserved after subsequent culture (Figure 1D). We did not observe any differences in the levels of CD16 expression between CD38WT and CD38KO NK cells (supplemental Figure 1B).
![Successful generation of CD38KO NK cells from ex vivo expanded PB-NK cells using Cas9/RNP. (A) Workflow for generating CD38KO NK cells from ex vivo expanded PB-NK cells using electroporation of Cas9/RNP. (B) CD38 expression in NK cells before and after Cas9/RNP-mediated CD38 deletion (n = 5; mean ± standard deviation [SD]). (C) Representative fluorescence-activated cell sorter (FACS) analyses of the purified CD38KO NK cells. Each figure indicates the percentage of CD38-expressing NK cells. Isotype controls are depicted with filled histograms. (D) Expansion of CD38WT and CD38KO NK cells.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/21/10.1182_blood.2020006200/1/m_bloodbld2020006200f1.png?Expires=1767705251&Signature=MmK-AWdiqXCu70vQXhA~i40IQnE5EX2o1aceS5aTEZCQa0GhG2ddcwSsrlAhfWI9WETlr~QIz0kLxzke-v2c6uIIEJbjJt5-w3U1xdxGrpSanklTnH29YXUrXXg7PfJI1TnM23gmBD77P814Hg-D9tukcyQvVGDx5Qd8tgpJ5k7ZiWPcqifVxGkD1iyCt3XerwSOfrLCkz6sJTUQuFNm-cfTxy0h-WdxbdjJ8t4o436EvpX0OMipymNcwhNEpdyqGXtfuxn8KjD6-MSK-PtJXHmoQhCsJgDZtdmDup0EwLj7SzcNCE7sioofJp4gZIS6QgSuBfk04jNbKMuj38KdzA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Successful generation of CD38KO NK cells from ex vivo expanded PB-NK cells using Cas9/RNP. (A) Workflow for generating CD38KO NK cells from ex vivo expanded PB-NK cells using electroporation of Cas9/RNP. (B) CD38 expression in NK cells before and after Cas9/RNP-mediated CD38 deletion (n = 5; mean ± standard deviation [SD]). (C) Representative fluorescence-activated cell sorter (FACS) analyses of the purified CD38KO NK cells. Each figure indicates the percentage of CD38-expressing NK cells. Isotype controls are depicted with filled histograms. (D) Expansion of CD38WT and CD38KO NK cells.
Successful generation of CD38KO NK cells from ex vivo expanded PB-NK cells using Cas9/RNP. (A) Workflow for generating CD38KO NK cells from ex vivo expanded PB-NK cells using electroporation of Cas9/RNP. (B) CD38 expression in NK cells before and after Cas9/RNP-mediated CD38 deletion (n = 5; mean ± standard deviation [SD]). (C) Representative fluorescence-activated cell sorter (FACS) analyses of the purified CD38KO NK cells. Each figure indicates the percentage of CD38-expressing NK cells. Isotype controls are depicted with filled histograms. (D) Expansion of CD38WT and CD38KO NK cells.
Low off-target effects of Cas9/RNP in NK cells
High-fidelity Cas9 has been shown to have low off-target editing because of its rapid degradation after electroporation.39 To study the off-target effects in CRISPR-modified NK cells, we performed WGS and found 26 genes with single nucleotide polymorphisms and indels exclusive to the CD38KO NK cells. Because we restricted our analysis to mutations in coding regions, all genes had mutations of moderate or high potential impact. In all, 18 genes had mutations categorized as moderate impact (missense and non-frameshift) and 8 genes (including CD38) had mutations categorized as high impact (startloss, stopgain, and frameshift) by SnpEff (supplemental Table 3). By RNA-seq, only 4 of the off-target genes with possible high-impact mutations are expressed at meaningful levels in NK cells (CC2D1B, DENND4B, KMT2C, and WDR89; supplemental Figure 2). These results show the efficiency and specificity of this guide RNA for CD38 targeting in NK cells.
Resistance of CD38KO NK cells to DARA-induced fratricide
It is likely that DARA induces NK cell fratricide via NK-to-NK ADCC by cross-linking CD38 and CD16.25 To determine whether CD38KO NK cells are resistant to DARA-induced fratricide, we evaluated conjugation and viability of paired CD38WT and CD38KO NK cells. DARA increased the formation of CD38WT NK cell conjugates but did not affect the formation of CD38KO NK cell conjugates (Figure 2A-B). Consistent with this result, DARA induced ADCC of CD38WT NK cells but not CD38KO NK cells (Figure 2C-D). NK cells engineered to lack CD16 (FCγRIII, CD16KO NK cells) showed no DARA-dependent ADCC or fratricide (supplemental Figure 3A-D). DARA showed no activity against MM cells in the absence of NK effector cells or complement (supplemental Figure 3C). The results presented here clearly delineate the contribution of NK effector function to DARA-dependent ADCC and DARA-dependent fratricide. They also show that CD16 and CD38 are necessary and sufficient for DARA-induced fratricide and that deletion of CD38 in NK cells renders them resistant to DARA-induced fratricide.
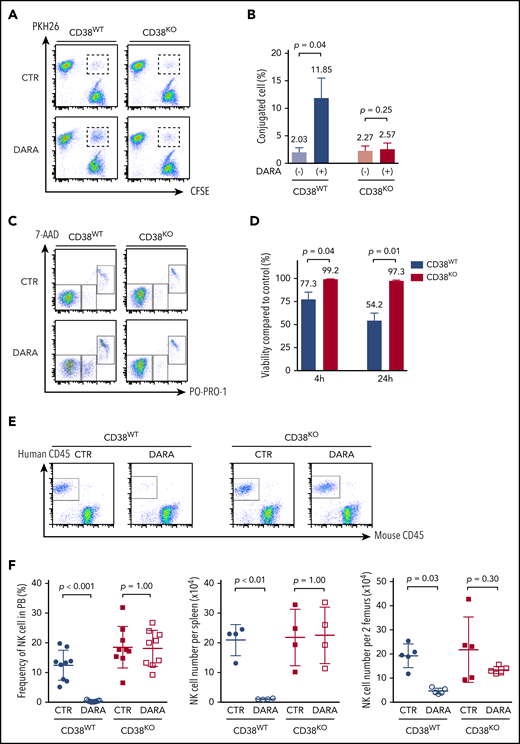
Resistance of CD38KO NK cells to DARA-induced fratricide. (A) Representative FACS analyses of the conjugation assay. (B) Summarized data of conjugation assays are shown (n = 3; mean ± SD). (C) Representative FACS analyses of the fratricide assay. (D) Viability of CD38WT and CD38KO NK cells treated with DARA compared with that of control (CTR) samples (n = 3; mean ± SD). (E) Representative FACS analyses of PB of NSG mice 7 days after treatment with DARA or saline. (F) Summarized data of NK cell persistence in NSG mice during treatment. The frequency of human NK cells in PB at day 7 and their absolute number in spleen and BM at day 9 are shown (n = 5; mean ± SD).
Resistance of CD38KO NK cells to DARA-induced fratricide. (A) Representative FACS analyses of the conjugation assay. (B) Summarized data of conjugation assays are shown (n = 3; mean ± SD). (C) Representative FACS analyses of the fratricide assay. (D) Viability of CD38WT and CD38KO NK cells treated with DARA compared with that of control (CTR) samples (n = 3; mean ± SD). (E) Representative FACS analyses of PB of NSG mice 7 days after treatment with DARA or saline. (F) Summarized data of NK cell persistence in NSG mice during treatment. The frequency of human NK cells in PB at day 7 and their absolute number in spleen and BM at day 9 are shown (n = 5; mean ± SD).
Next, to determine whether the resistance of CD38KO NK cells to DARA would also contribute to superior persistence in vivo, we infused CD38WT or CD38KO NK cells into NSG mice treated with DARA and examined NK cell frequency in PB, spleen, and BM. CD38WT and CD38KO NK cells showed comparable engraftment in control mice (Figure 2E-F). In contrast, treatment with DARA significantly reduced engraftment of CD38WT NK cells but had no effect on persistence of CD38KO NK cells (Figure 2E-F). CD38WT NK cells were depleted by DARA in spleen and BM as well as in PB, whereas CD38KO NK cells showed no significant depletion in any of these compartments between control and DARA-treated mice (Figure 2F). Taken together, these results show that CD38KO cells are resistant to DARA in vitro and in vivo.
Superior DARA-mediated ADCC of CD38KO NK cells against MM cells
Because DARA-induced depletion of NK cells would blunt ADCC against target cells,23 we hypothesized that CD38KO NK cells would also kill target cells more efficiently than CD38WT NK cells. To study this, we tested the cytotoxicity of paired CD38WT and CD38KO NK cells in the presence or absence of DARA against different MM cell lines with high, low, or no levels of CD38 expression (Figure 3A). The direct cytotoxicity against each MM cell line was equivalent between CD38WT and CD38KO NK cells; however, in the presence of DARA, CD38KO NK cells showed significantly higher cytotoxicity against CD38+ target cells (Figure 3B). Because we observed no DARA-mediated lysis in the absence of NK effector cells (supplemental Figure 4), the superior killing efficiency of CD38KO NK cells is attributed to their higher ADCC activity (Figure 3C). As expected, neither CD38WT nor CD38KO NK cells exhibited ADCC against the CD38– cell line U266. Intriguingly, CD38WT NK cells showed marginal or no ADCC against MM cells with low levels of CD38 expression such as OPM-2 and KMS-11, whereas CD38KO NK cells demonstrated significantly stronger ADCC against these MM cell lines. Similar to the results with cell lines, CD38KO NK cells showed higher DARA-mediated ADCC activity against primary MM samples (Figure 3D-E), including improved cytotoxicity against primary CD38low MM cells from a DARA-resistant patient (Figure 3F; supplemental Figure 5). When analyzed across MM target cell lines and primary samples, the relative benefit of CD38KO NK cells over paired CD38WT NK cells was inversely correlated with the CD38 expression level on the target cells (r = –0.786; P = .048; supplemental Figure 6). Thus, our results suggest that CD38KO NK cells may improve the efficacy of DARA against R/R MM that is otherwise DARA resistant because of low CD38 expression.
![Enhanced DARA-mediated ADCC activity of CD38KO NK cells against MM cell lines and primary MM cells. (A) Representative FACS analyses of CD38 expression of NK cells and myeloma cell lines. Each panel shows the mean fluorescence intensity (MFI). Isotype controls are depicted with filled histograms. (B-C) Representative data of cytotoxicity and DARA-mediated ADCC activity of paired CD38WT and CD38KO NK cells against myeloma cell lines and (D) a representative primary MM sample. (E) ADCC activity of paired CD38WT and CD38KO NK cells against primary MM samples (effector-to-target [E:T] ratio is 0.1:1) (F) Cytotoxicity of paired CD38WT and CD38KO NK cells against primary DARA-resistant MM cells in the presence of DARA.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/21/10.1182_blood.2020006200/1/m_bloodbld2020006200f3.png?Expires=1767705251&Signature=I2C-7eM-q~ZrdpGbxkcrgf60Llb7pKFCqiTjUe2KLP2z~grjxpsJEJOLpNAbmlAbHjXutMYifyn2K1Ikw8kquijS~LiLlVry8bb-goagLf2vlZ~leM5HTN6zhJSMLy9DE6ti485ha7aumJ0mprMDZuHC6rymghOuxs~jBDOg-PwnQHMmNvel~vkYC4TMhi5w8jchqUd6OkqcCKjZgkfmasXwLMrtaA6akKGk4Ymg4Obwzn2Ymmj8aBRB9fxNncGkOIifyhRy2EIc786fNwcqgswEwvnvaapYLQO6Zb2AuH5tBNrJ6abfndV6F7SDsi2rXLE~vVvdKRQkPcwzF9OnQg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Enhanced DARA-mediated ADCC activity of CD38KO NK cells against MM cell lines and primary MM cells. (A) Representative FACS analyses of CD38 expression of NK cells and myeloma cell lines. Each panel shows the mean fluorescence intensity (MFI). Isotype controls are depicted with filled histograms. (B-C) Representative data of cytotoxicity and DARA-mediated ADCC activity of paired CD38WT and CD38KO NK cells against myeloma cell lines and (D) a representative primary MM sample. (E) ADCC activity of paired CD38WT and CD38KO NK cells against primary MM samples (effector-to-target [E:T] ratio is 0.1:1) (F) Cytotoxicity of paired CD38WT and CD38KO NK cells against primary DARA-resistant MM cells in the presence of DARA.
Enhanced DARA-mediated ADCC activity of CD38KO NK cells against MM cell lines and primary MM cells. (A) Representative FACS analyses of CD38 expression of NK cells and myeloma cell lines. Each panel shows the mean fluorescence intensity (MFI). Isotype controls are depicted with filled histograms. (B-C) Representative data of cytotoxicity and DARA-mediated ADCC activity of paired CD38WT and CD38KO NK cells against myeloma cell lines and (D) a representative primary MM sample. (E) ADCC activity of paired CD38WT and CD38KO NK cells against primary MM samples (effector-to-target [E:T] ratio is 0.1:1) (F) Cytotoxicity of paired CD38WT and CD38KO NK cells against primary DARA-resistant MM cells in the presence of DARA.
Inhibitory effect of ATRA on NK cell cytotoxicity
Downregulation of CD38 on MM cells is considered to play an important role in developing resistance to DARA.40,41 Treatment with ATRA can overcome this resistance through upregulation of CD38 levels on CD38low target cells.23 To investigate potential synergy in upregulating CD38 on MM cells and deleting CD38 on NK cells, we pretreated MM cells with ATRA before assessing ADCC with DARA and NK cells. We confirmed that pretreatment with ATRA upregulates CD38 levels on MM target cells (supplemental Figure 7) and showed that it improves DARA-mediated ADCC in the presence of both CD38WT and paired CD38KO NK cells (Figure 4A-B). We then examined the direct effects of ATRA on CD38 expression on NK cells in vivo using PB-NK cells from patients with acute promyelocytic leukemia treated with ATRA during consolidation therapy. Compared with samples obtained before treatment, treatment with ATRA was associated with significant upregulation of CD38 on the PB-NK cells of these patients (Figure 4C). Similarly, ex vivo treatment with ATRA upregulates CD38 levels on CD38WT but not on the paired CD38KO NK cells (Figure 4D). In addition, ATRA also enhanced fratricide of CD38WT NK cells (Figure 4E). In contrast to pretreatment of MM cells with ATRA, concurrent treatment of both MM and NK cells with ATRA significantly impaired the DARA-mediated ADCC of CD38WT NK cells but had no impact on the DARA-mediated ADCC of CD38KO NK cells (Figure 4F). Interestingly, ATRA significantly reduced direct cytotoxicity of both CD38WT and the paired CD38KO NK cells to the same extent (Figure 4F). Thus, ATRA-induced upregulation of CD38 on MM target cells may be offset by increased NK cell fratricide and impaired NK cell function, decreasing the overall efficacy of DARA, which can be mitigated by the use of CD38KO NK cells (Figure 4G).
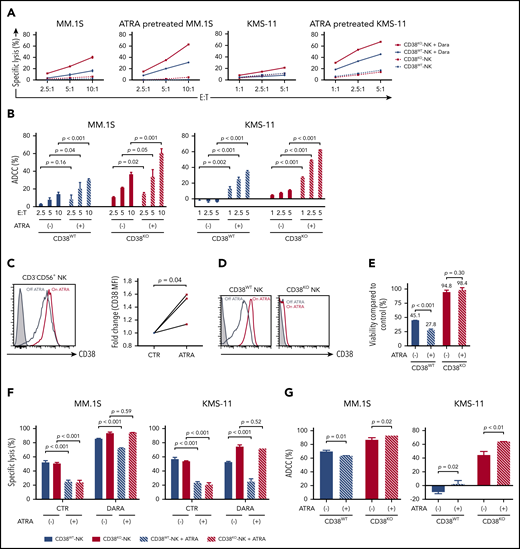
Inhibitory effects of ATRA on DARA-mediated NK cell cytotoxicity. (A-B) Cytotoxicity and DARA-mediated ADCC activity of paired CD38WT and CD38KO NK cells against myeloma cell lines pretreated with 50 nM ATRA for 48 hours (mean ± SD). (C) Left panel shows representative FACS analyses data for CD38 expression on NK cells (CD3–CD56+) from patients during ATRA treatment or no therapy. Frozen PB mononuclear cells were thawed and analyzed at once. Right panel shows fold increase of MFI (CD38) of NK cells during ATRA therapy compared with no therapy for 3 different patients. (D) Representative FACS analyses data of CD38 expression on CD38WT and CD38KO NK cells 48 hours after incubation with 50 nM ATRA or solvent control. Control and ATRA-treated samples are shown with steel blue and red lines, respectively. Unstained controls are depicted with filled histograms. (E) Viability of CD38WT and CD38KO NK cells treated with DARA for 48 hours in the presence of 50 nM ATRA or solvent control compared with that of control samples (mean ± SD). (F-G) Cytotoxicity and DARA-mediated ADCC activity of paired CD38WT and CD38KO NK cells against myeloma cell lines in a 48-hour cytotoxicity assay in the presence of 50 nM ATRA or solvent control. E:T ratio is 0.25:1 for MM.1S and 0.5:1 for KMS-11 (mean ± SD).
Inhibitory effects of ATRA on DARA-mediated NK cell cytotoxicity. (A-B) Cytotoxicity and DARA-mediated ADCC activity of paired CD38WT and CD38KO NK cells against myeloma cell lines pretreated with 50 nM ATRA for 48 hours (mean ± SD). (C) Left panel shows representative FACS analyses data for CD38 expression on NK cells (CD3–CD56+) from patients during ATRA treatment or no therapy. Frozen PB mononuclear cells were thawed and analyzed at once. Right panel shows fold increase of MFI (CD38) of NK cells during ATRA therapy compared with no therapy for 3 different patients. (D) Representative FACS analyses data of CD38 expression on CD38WT and CD38KO NK cells 48 hours after incubation with 50 nM ATRA or solvent control. Control and ATRA-treated samples are shown with steel blue and red lines, respectively. Unstained controls are depicted with filled histograms. (E) Viability of CD38WT and CD38KO NK cells treated with DARA for 48 hours in the presence of 50 nM ATRA or solvent control compared with that of control samples (mean ± SD). (F-G) Cytotoxicity and DARA-mediated ADCC activity of paired CD38WT and CD38KO NK cells against myeloma cell lines in a 48-hour cytotoxicity assay in the presence of 50 nM ATRA or solvent control. E:T ratio is 0.25:1 for MM.1S and 0.5:1 for KMS-11 (mean ± SD).
Higher OXPHOS activity in CD38KO NK cells
CD38 is a 46-kDa type II transmembrane glycoprotein and has been shown to have multiple functions including ectoenzymatic activity as an NAD+ hydrolase to regulate intracellular NAD+ levels.10 Because NAD+ is an essential cofactor for enzyme-catalyzed reactions that contributes to adenosine triphosphate (ATP) production, CD38 plays an important role in cellular metabolism.30 A recent study reported that CD38 knockout in T cells results in higher levels of intracellular NAD+, which fuels OXPHOS and ATP synthesis, and consequently leads to higher cytotoxicity against cancers.42 Metabolic commitment has a crucial role in cytotoxicity and survival of NK cells in the tumor environment similar to that of T cells.43 These findings prompted us to investigate the impact of CD38 knockout on NK cell metabolism. First, we performed RNA-seq on WT and CD38KO NK cells, identified DEGs, and used IPA to identify differentially regulated pathways from the DEGs. IPA showed a significant change in cholesterol biosynthesis (P < .00001) and OXPHOS (P < .00001) pathways in CD38KO NK cells. Analysis of genes in those pathways identified a modest but significant increase in expression of mitochondrial genes specifically associated with ATP synthesis, NAD recycling, and electron transport in CD38KO NK cells (Table 1; Figure 5A; supplemental Figure 8). Principle component analysis revealed significant donor-dependent variation at baseline but a consistent directional change in response to CD38 deletion among all donor pairs, except for donor #11 which was already at the far end of the spectrum at baseline (Figure 5B). Considering the upregulation of genes in these metabolic pathways, we next examined the cellular metabolism of CD38KO NK cells. By using a mitochondrial stress test assay, we observed higher OCR and comparable ECAR, resulting in a significantly higher OCR:ECAR ratio in CD38KO NK cells compared with CD38WT NK cells (Figure 5C-D). This result suggests that deletion of CD38 induces NK cells to preferentially use OXPHOS to achieve their bioenergetic demands. Importantly, CD38KO NK cells also had higher spare respiratory capacity and mitochondrial respiratory capacity compared with CD38WT NK cells (Figure 5D). These favorable metabolic shifts are consistent with the enhanced DARA-mediated cytotoxicity of CD38KO NK cells.
Fold changes and P values of DEGs associated with significantly altered metabolic pathways identified by IPA
| Gene | Expression fold change | P |
|---|---|---|
| ATP5MF | 1.258 | .0323 |
| ATP5PB | 1.132 | .0471 |
| ATP5PF | 1.211 | .0492 |
| COX11 | 1.129 | .00759 |
| COX6A1 | 1.178 | .0247 |
| COX7A2 | 1.175 | .00928 |
| COX7B | 1.118 | .0362 |
| NDUFA4 | 1.173 | .0125 |
| UQCR10 | 1.181 | .0456 |
| UQCRC2 | 1.144 | .0366 |
| HMGCR | 1.089 | .0255 |
| HSD17B7 | 1.123 | .036 |
| IDI1 | 1.113 | .0259 |
| LBR | 1.102 | .0362 |
| MSMO1 | 1.124 | .00151 |
| NSDH1 | 1.135 | .0413 |
| Gene | Expression fold change | P |
|---|---|---|
| ATP5MF | 1.258 | .0323 |
| ATP5PB | 1.132 | .0471 |
| ATP5PF | 1.211 | .0492 |
| COX11 | 1.129 | .00759 |
| COX6A1 | 1.178 | .0247 |
| COX7A2 | 1.175 | .00928 |
| COX7B | 1.118 | .0362 |
| NDUFA4 | 1.173 | .0125 |
| UQCR10 | 1.181 | .0456 |
| UQCRC2 | 1.144 | .0366 |
| HMGCR | 1.089 | .0255 |
| HSD17B7 | 1.123 | .036 |
| IDI1 | 1.113 | .0259 |
| LBR | 1.102 | .0362 |
| MSMO1 | 1.124 | .00151 |
| NSDH1 | 1.135 | .0413 |

Favorable metabolic reprogramming of CD38KO NK cells. (A) Heat map of DEGs of significantly altered pathways (cholesterol biosynthesis and OXPHOS) as determined by IPA, based on normalized RNA-seq data of paired CD38WT and CD38KO NK cells (n = 6). (B) Principle components analysis (PCA) of DEGs, showing consistent effect of CD38 deletion for each donor despite wide interdonor variability. (C) Summarized data of metabolic analysis of paired CD38WT and CD38KO NK cells (n = 3; mean ± SD). (D) Graphical analysis of basal OCR, ECAR, OCR/ECAR, and spare respiratory capacity (SRC) derived from (C). All experiments were achieved using quintuplicate samples. FCCP, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone; ROT/AA, rotenone and antimycin A.
Favorable metabolic reprogramming of CD38KO NK cells. (A) Heat map of DEGs of significantly altered pathways (cholesterol biosynthesis and OXPHOS) as determined by IPA, based on normalized RNA-seq data of paired CD38WT and CD38KO NK cells (n = 6). (B) Principle components analysis (PCA) of DEGs, showing consistent effect of CD38 deletion for each donor despite wide interdonor variability. (C) Summarized data of metabolic analysis of paired CD38WT and CD38KO NK cells (n = 3; mean ± SD). (D) Graphical analysis of basal OCR, ECAR, OCR/ECAR, and spare respiratory capacity (SRC) derived from (C). All experiments were achieved using quintuplicate samples. FCCP, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone; ROT/AA, rotenone and antimycin A.
Discussion
The use of DARA has been effective in patients with R/R MM. However, despite being incorporated into first-line regimens and the acceptance of DARA as standard of care in the treatment of MM,44 it is increasingly clear that disease relapse is inevitable.12 Resistance mechanisms of MM cells to treatment with DARA are beginning to be elucidated.40,41 Some of these mechanisms may overlap with previously proposed mechanisms of resistance to IMiDs or proteasome inhibitors, such as tumor heterogeneity and the role of the BM microenvironment,45,46 but other mechanisms may be unique to monoclonal antibody therapies.
Monoclonal antibodies eliminate targets through 4 mechanisms: CDC, ADCC, ADCP, and activation-induced cell death through receptor cross-linking. Thus, resistance occurs through suppression of these mechanisms. For instance, DARA resistance has been associated with overexpression of complement inhibitory proteins (CD55 and CD59) on the surface of MM cells, impairing DARA-mediated CDC.19 With respect to ADCC, a unique situation occurs with DARA in that NK cells expressing high levels of CD38 are eliminated during treatment, which cripples DARA-mediated ADCC. Rescue of DARA-mediated ADCC by adoptive transfer of ex vivo expanded NK cells was successful in a preclinical model25 using CD38low NK cells. However, these CD38low NK cells re-acquired CD38 expression during ex vivo expansion and thus regained susceptibility to DARA-mediated elimination, making clinical translation unlikely. In addition, patients with MM, particularly those treated with DARA, have relatively low numbers of immune cells, including NK cells.11 Thus, we focused our studies on allogeneic NK cells in which a third-party universal donor strategy would allow the selection of donors with desirable killer cell immunoglobulin-like receptor (KIR) genotypes or FCγRIIIA polymorphisms for optimal NK cell function.
Here, we generated CD38KO NK cells by using the CRISPR/Cas9 system. These cells were resistant to DARA-induced conjugation and fratricide and persisted in the presence of DARA in vivo. CD38KO NK cells showed superior ADCC activity against MM cell lines and primary samples when compared with the paired CD38WT cells. Interestingly, CD38KO NK cells were particularly effective against MM cell lines with low CD38 expression and MM cells from a patient who relapsed during DARA treatment, whereas CD38WT NK cells had minimal to no activity against those target cells. Given the selective pressure for low CD38-expressing MM cells during treatment with DARA,19 CD38KO NK cells could reinforce the therapeutic effect of DARA against this residual disease.
Deletion of CD38 on NK cells was associated with increased mitochondrial respiratory capacity of these cells and a compensatory transcriptomic profile favoring OXPHOS metabolism and cholesterol synthesis. Although we did not directly investigate the role of this metabolic shift in effector function of CD38KO NK cells, previous studies showed that knocking out CD38 in T cells results in higher OXPHOS activity and antitumor effect.42 Further investigation is needed to identify the mechanisms by which CD38 deletion alters NK cell metabolism, although others have shown that increased NAD+ levels correlate with increased adenosine diphosphate ribose and adenosine in hypoxic microenvironments.47
Antigen density is an important factor in target recognition and effector function by monoclonal antibodies, and thus upregulation of CD38 levels on target cells has the potential to enhance DARA activity. Recently, the clinical synergy between IMiDs and DARA has been attributed in part to the upregulation of CD38 on MM cells by IMiDs.18 ATRA was also shown to upregulate CD38 levels on MM cells and sensitize them to DARA-mediated CDC and ADCC.23 As opposed to IMiDs that have a known favorable immunomodulatory profile, little is known about the effects of ATRA on the ADCC activity of NK cells.48,49 Here, we confirmed that treatment with ATRA upregulates CD38 levels on MM cells but also showed that it upregulates CD38 on ex vivo expanded NK cells and on in vivo circulating NK cells of patients with acute promyelocytic leukemia. This modulatory effect on NK cells may enhance DARA-induced fratricide and impair DARA-mediated ADCC against MM cells in vivo. Although CD38KO NK cells were free from fratricide, direct cytotoxicity of both CD38WT and CD38KO NK cell was significantly suppressed by ATRA. Thus, ATRA-mediated upregulation of CD38 levels on MM cells was offset by its negative impact on NK cell function. Taken together, the net result of treatment with ATRA was overall impaired DARA-mediated ADCC activity of CD38WT NK cells, whereas the use of CD38KO NK cells rescued the negative impact of ATRA on ADCC.
It is important to mention that treatment with ATRA may boost DARA-mediated CDC via decreasing CD55 and CD59 expression on MM cells.19,23 Even though most monoclonal antibodies for clinical development are chosen based on their CDC activity,50-52 it is unclear which of the 4 mechanisms of action plays the most important role in the clinical settings.53-56 Preclinical models to differentiate these mechanisms have been difficult because of the high efficacy of DARA alone in eradicating MM in murine xenografts, such that the addition of ex vivo expanded NK cells has a relatively small benefit over DARA alone.25 Development of CD38low MM patient-derived xenografts may be useful for pharmacokinetic modeling of DARA and NK cell combinations and in comparing CD38KO and CD38WT NK cells. The current clinical trial (NCT02751255) testing the combination of ATRA and DARA for patients with MM uses a unique therapeutic schedule that provides transient and staggered exposure to these drugs. The pharmacokinetics of DARA is relatively constant during treatment,57,58 and ATRA levels are strictly regulated by complex systemic and tissue-dependent feedback mechanisms.59,60 In addition to the clinical outcomes using this combination, it is of interest to understand the impact of this treatment schedule on NK cell numbers, functions, and DARA-mediated ADCC and CDC. In addition, it has been shown that anti-KIR antibodies enhance DARA-mediated lysis of primary myeloma cells.61 Investigating the impact of KIR function and genotype and FCγRIIIA polymorphisms on the ADCC activity of CD38KO and CD38WT NK cells is an important area of future research and a necessary step toward clinical translation of these findings.
In this study, we have provided proof of concept that CD38KO NK cells could boost the effects of DARA against MM cells. Our newly developed DNA-free approach using Cas9/RNP succeeded in efficient generation of CD38KO NK cells with very low frequency of off-target effects as assessed by WGS.
We also applied previously published methods here to expanding genetically modified NK cells, which is critical for achieving clinically significant numbers.62 Collectively, our results demonstrate the rationality and feasibility of ex vivo expanded CD38KO NK cells for adoptive immune therapy in combination with DARA against MM but also potentially other CD38+ hematologic malignancies such as acute myeloid leukemia,63 and B- and T-cell lymphoblastic leukemias64,65 and lymphomas.66,67 In addition, our method for CD38 deletion in NK cells may be applied to generating CD38 chimeric antigen receptor NK cells to avoid their fratricide during cell manufacturing and effective antitumor activity in vivo.68
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Contact the corresponding author for original data.
The online version of this article contains a data supplement.
Acknowledgments
This work is supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (K08 HL127269 and R03 HL145226).
Authorship
Contribution: M.N.K., Y.N., E.E., M.d.S.F.P., and D.W. performed the research; S.A.A., P.H.I., and I.M.B. provided vital reagents; and M.N.K., Y.N., D.A.L., and G.G. designed the research, analyzed the data, and wrote the article.
Conflict-of-interest disclosure: D.A.L. has consulting relationships with Caribou Biosciences and Courier Therapeutics, and equity/leadership/consulting relationships with Kiadis Pharma Netherlands B.V. The remaining authors declare no competing financial interests.
Correspondence: Gabriel Ghiaur, Johns Hopkins University, 1650 Orleans St, CRB I, Room 243, Baltimore, MD 21287; e-mail: gghiaur1@jhmi.edu.
