

Key Points
Coronavirus-specific polyfunctional T cells can be expanded from convalescent individuals for use for patients after bone marrow transplant.
SARS-CoV-2 T-cell products target structural viral proteins, including commonly recognized regions in the C terminus of membrane protein.

Abstract
T-cell responses to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) have been described in recovered patients, and may be important for immunity following infection and vaccination as well as for the development of an adoptive immunotherapy for the treatment of immunocompromised individuals. In this report, we demonstrate that SARS-CoV-2–specific T cells can be expanded from convalescent donors and recognize immunodominant viral epitopes in conserved regions of membrane, spike, and nucleocapsid. Following in vitro expansion using a good manufacturing practice-compliant methodology (designed to allow the rapid translation of this novel SARS-CoV-2 T-cell therapy to the clinic), membrane, spike, and nucleocapsid peptides elicited interferon-γ production, in 27 (59%), 12 (26%), and 10 (22%) convalescent donors (respectively), as well as in 2 of 15 unexposed controls. We identified multiple polyfunctional CD4-restricted T-cell epitopes within a highly conserved region of membrane protein, which induced polyfunctional T-cell responses, which may be critical for the development of effective vaccine and T-cell therapies. Hence, our study shows that SARS-CoV-2 directed T-cell immunotherapy targeting structural proteins, most importantly membrane protein, should be feasible for the prevention or early treatment of SARS-CoV-2 infection in immunocompromised patients with blood disorders or after bone marrow transplantation to achieve antiviral control while mitigating uncontrolled inflammation.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a novel coronavirus first reported in December 2019 from Wuhan, China, is responsible for the ongoing pandemic of coronavirus disease 2019 (COVID-19).1 The adaptive immune response to SARS-CoV-2 remains ill-defined and there is an urgent need to fill this gap in knowledge to enable the development of effective vaccines and therapies. Antibody responses to the spike and nucleocapsid proteins are well described,2,3 and recently the characterization of T-cell responses to SARS-CoV-2 predominantly to spike, membrane, and nucleocapsid proteins has also been reported.4-11 Recent studies have reported that both CD4+ and CD8+ T-cell responses to SARS-CoV-2 are detectable in convalescent patients, as well as in a proportion of unexposed individuals, albeit at lower levels. Recent reports have also suggested that immunocompromised patients may be at high risk of severe and potentially prolonged disease, suggesting that T-cell immunity is essential for overcoming COVID-19.12,13 Studies of the related virus SARS-CoV demonstrated that T cells recognizing viral epitopes within SARS-CoV structural proteins were integral in viral clearance, and remained detectable for >10 years after exposure.14,15
Knowledge of T-cell epitopes recognized in other viruses such as Epstein-Barr virus (EBV), cytomegalovirus (CMV), and adenovirus have successfully led to the development of adoptive immunotherapy with ex vivo expanded virus-specific T cells (VSTs). This approach has been highly successful in preventing or treating viral infections in high-risk patients after bone marrow transplant (BMT) with minimal risk of graft-versus-host disease.16,17 To date, >1000 patients have been treated internationally in phase 1/2 protocols using VSTs.18-24 Importantly, expansion of VSTs in vivo correlates strongly with antiviral efficacy.23,25,26 Hence, the expansion of such approaches to include SARS-CoV-2–specific T cells may also offer protection from COVID-19 to these vulnerable individuals. Here, we define the immunodominant T-cell epitopes within conserved regions of SARS-CoV-2 structural proteins, including the novel discovery that SARS-CoV-2–specific T cells predominantly recognize regions in the C terminus of the membrane protein, which represents a critical “hot spot” for CD4-restricted T-cell epitopes. We also noted an association between SARS-CoV-2 seropositivity and the breadth of T-cell responses to structural viral proteins in patients who recover from COVID-19. These data suggest that patients who mount an antibody response to SARS-CoV-2 are more likely to have a broader T-cell response following COVID-19, which may have implications for protective immunity in recovered patients. It also provides proof of concept for optimal donor section for the rapid manufacture of good manufacturing practice (GMP)-compliant SARS-CoV-2–specific T-cell therapeutics, with the potential to prevent or treat COVID-19 in immunocompromised patients with blood disorders and/or after BMT.
Methods
Donors
Peripheral blood mononuclear cells (PBMCs) from volunteers, both healthy and those with presumed or documented COVID-19 infection, were obtained from Children’s National Hospital (Washington, DC) and the National Institutes of Health under informed consent approved by the Institutional Review Board of both institutions in accordance with the Declaration of Helsinki.
Generation of SARS-CoV-2–specific T cells
Evaluated T-cell products included SARS-CoV-2–specific T cells (CSTs), manufactured from PBMCs of seropositive and seronegative volunteers. VSTs were produced using a rapid expansion protocol previously described. Briefly, PBMCs were pulsed with a mix of overlapping peptide pools encompassing viral structural proteins (1 μg/antigen per 15 × 106 PBMCs) for 30 minutes at 37°C. Peptide libraries of 15-mers with 11 amino acid overlaps encompassing the spike, membrane, nucleocapsid, and envelope proteins were generated (A&A Peptide, San Diego, CA) from the SARS-CoV-2 reference sequence (NC_045512.2), and were pooled equally by mass and reconstituted to a working concentration of 1 μg/μL. After incubation, cells were resuspended with interleukin-4 (IL-4; 400 IU/mL; R&D Systems, Minneapolis, MN) and IL-7 (10 ng/mL; R&D Systems) in CTL media consisting of 45% RPMI (GE Healthcare, Logan, UT), 45% Click medium (Irvine Scientific, Santa Ana, CA), 10% fetal bovine serum (FBS), and supplemented with 2 mM GlutaMax (Gibco, Grand Island, NY) according to our GMP-compliant standard operating procedures. Cytokines were replenished on day 7. On day 10, cells were harvested and evaluated for antigen specificity and functionality. A subset of samples was restimulated with autologous PBMCs that were pulsed with the viral peptide libraries, irradiated at 75 Gy, and cocultured with the CSTs at a ratio of 1:4 (CSTs to PBMCs). These restimulated cells were incubated in IL-4 (400 IU/mL) and IL-7 (10 ng/mL), with cytokines replenished at day 17, and harvested at day 21 for further testing.
Isolation and maintenance of SARS-CoV-2–specific T-cell clones
Membrane and spike-specific T cells were isolated from frozen VSTs using an interferon-γ (IFN-γ) capture assay protocol previously described. Briefly, VSTs were thawed, washed in warm X-VIVO-15, and resuspended at a concentration of 1 × 107 cells/mL. VSTs were stimulated for 3 hours with overlapping peptide pools encompassing viral antigens to spike and membrane to a final concentration of 1 μg/mL. T cells producing IFN-γ in response to this stimulation were enriched using the IFN-γ Sectretion Detection and Enrichemnt Kit (130-054-201; Miltenyi Biotec, Bergisch Gladbach, Germany) in accordance with the manufacturer’s instructions. These T cells were plated at a series of dilutions in 96-well plates with irradiated feeder medium (RPMI 1640 supplemented with 10% FBS, l-glutamine, and PenStrep [R-10]) with 1 × 106 cells/mL 5000 rad irradiated PBMC + 50 U/mL IL-2 + 10 ng/mL IL-15 + 0.1 μg/mL each of anti-CD3 (Ultra-LEAF purified anti-human CD3 antibody clone OKT3; BioLegend, San Diego, CA) and anti-CD28 (Ultra-LEAF purified Anti-human CD28 antibody clone 28.2; BioLegend). Membrane and spike-specific T-cell clones were expanded biweekly with irradiated feeder medium. One month later, colonies were selected from the lowest dilution plates with positive wells (<1/3 of wells positive) and screened for responsiveness to membrane or spike peptide pools by intracellular cytokine staining for IFN-γ and tumor necrosis factor-α (TNF-α).
IFN-γ ELISpot assay
Antigen specificity of T cells was measured by IFN-γ enzyme-linked immunospot (ELISpot; Millipore, Burlington, MA). T cells were plated at 1 × 105/well with no peptide, actin (control), or each of the individual SARS-CoV-2 pepmixes (200 ng per peptide per well). Plates were sent for IFN-γ spot-forming cells counting (Zellnet Consulting, Fort Lee, NJ).
Flow cytometry
VSTs were stained with fluorophore-conjugated antibodies against CD4, CD8, TCRαβ, TCRγδ, CXCR3, CXCR5, CCR6, CD127, CD25, and CD56 (Miltenyi Biotec; BioLegend). All samples were acquired on a CytoFLEX cytometer (Beckman Coulter, Brea, CA). Intracellular cytokine staining was performed as follows: 1 × 106 VSTs were plated in a 96-well plate and stimulated with pooled pepmixes or individual peptides (200 ng per peptide per well) or actin (control) in the presence of brefeldin A (Golgiplug; BD Biosciences, San Jose, CA) and CD28/CD49d (BD Biosciences) for 6 hours. T-cells were fixed, permeabilized with Cytofix/Cytoperm solution (BD Biosciences), and stained with IFN-γ and TNF-α and IL-2 antibodies (Miltenyi Biotec).
For intracellular flow cytometry of T-cell clones, cells were stimulated with membrane and spike peptide pools to a concentration of 1 μg/mL, and incubated at 37°C 5% CO2. After 1 hour, 1 μg/mL of brefeldin A was added to each well, and plates were incubated for another 5 hours. Cells were then washed in 2% FBS phosphate-buffered saline and surface stained with fluorochrome-conjugated antibodies to CD3-Brilliant Violet 785 clone OKT3, CD4-Alexa Fluor 700 clone RPA-T4, CD8-FITC clone RPA-T8, OX40-Brilliant Violet 711 clone Ber-ACT35 (ACT35) (all from BioLegend), CD69-APC-eFluor 780 clone FN50, and Fixable Aqua Viability Dye (both from Invitrogen). Cells were fixed, permeabilized using BD Cytofix/Cytoperm solution and stained with anti-IFN-γ Brilliant Violet 421 clone 4S.B3, anti-TNF-α PerCP-Cyanine5.5 clone Mab11 (both from BioLegend). Cells were analyzed on an Attune NxT flow cytometer. Data were analyzed with FlowJo X (FlowJo LLC, Ashland, OR).
Epitope mapping
CSTs were tested for specificity to minipools containing 8 to 24 peptides spanning the SARS-CoV2 antigens by IFN-γ ELISpot. Cross-reactive pools were analyzed and individual peptides were tested to confirm epitope specificity. In silico predictions of major histocompatibility complex (MHC) restrictions was performed using MARIA (http://maria.stanford.edu) and NetMHCIIPan (http://www.cbs.dtu.dk/services/NetMHCIIpan-4.0/).27,28
MHC restrictions were narrowed through use of blocking antibodies targeting MHC class II proteins. Briefly, CSTs were incubated were pulsed with 1 μg/mL of spike or membrane peptide pools and blocked with 10 μg/mL of either anti-HLA-DR, anti-HLA-DQ, or anti-HLA-DR,DP,DQ (BioLegend) for 30 minutes. Cells were washed 3 times with R10, and then blocked again with the same concentration of antibodies. After 1 hour, 1 μg/mL of brefeldin A was added to each well, and plates were incubated for another 5 hours. Cells were then washed and stained for surface markers and intracellular cytokines as described previously.
To confirm the restricted HLA allele, CSTs were plated at 1 × 105 per well with partially HLA-matched phytohemagglutinin-treated lymphoblasts (phytohemagglutinin blasts, 25 Gy irradiated) either alone or pulsed with peptide (1 μg/mL), and tested via IFN-γ ELISpot.
Luciferase immunoprecipitation systems for measurement of SARS-CoV-2 antibodies
Testing for antibodies to spike and nucleocapsid proteins were performed using a luciferase immunoprecipitation system assay, as recently described.3 Briefly, plasma samples were incubated with spike and nucleocapsid proteins fused to Gaussia and Renilla luciferase, respectively, protein A/G beads were added, the mixture was washed, coelenterazine substrate (Promega) was added, and luciferase activity was measured in light units with a Berthold 165 LB 960 Centro Microplate Luminometer. Antibody levels were reported as the geometric mean level with 95% confidence interval. Cutoff limits for determining positive antibodies in the SARS-CoV-2–infected samples were based on the mean plus 3 standard deviations of the serum values derived from uninfected blood donor controls or by receiver operator characteristics analysis. For some of the data percentages for categorical variables, mean and range, geometric mean, and 95% confidence interval were used to describe the data. Wilcoxon signed-rank tests were used for statistical analysis.
Multiplex cytokine assay
CSTs were plated at 1 × 105 per well in 96-well plates, stimulated with pooled pepmixes (200 ng/peptide/well) or control actin peptide, and incubated 48 hours. Supernatants were harvested and the cytokine profile analysis was performed using the Bio-plex Pro Human 17-Plex Cytokine Assay Kit (Bio-Rad, Hercules, CA), and read on a MAGPIX system (Luminex, Austin, TX).
Chromium release assay
Phytohemagglutinin blasts were labeled with chromium-51 (Perkin Elmer, Waltham, MA) at 10 μCi per 5 × 105 cells. CST were coplated with 51Cr-labeled, MHC-mismatched irradiated phytohemagglutinin blasts at effector:target ratios between 40:1 and 5:1, and incubated at 37°C for 4 hours. Maximal release was evaluated by lysis of 51Cr-labeled targets with Triton-X-100. Supernatants were transferred to lumiplates and read on a MicroBeta2 Plate Reader (Perkin Elmer). Specific lysis was calculated as follows: (experimental counts per minute [CPM] – background CPM)/(maximal CPM – background CPM).
Statistical analysis
Statistical analysis was performed in SAS (SAS Institute, Cary, NC). Pearson/Speakman calculations were used for correlations of T-cell and antibody responses of individual antigens, and Pearson χ2 test was used for binary correlations of T-cell and antibody responses. Graphs were produced in Prism (GraphPad, San Diego, CA). Immunodominance was defined as antigens and/or epitopes that induce statistically significant responses on IFN-γ ELISpot and/or intracellular cytokine staining in comparison with control peptides, and are recognized by multiple individuals.
Results
The majority of convalescent patients show antibody responses to SARS-CoV-2
Forty-six convalescent donors from the eastern and midwestern United States with presumptive recent COVID-19 (36 polymerase chain reaction [PCR]-proven and 10 presumed positive because they were: (1) symptomatic and in close contact with PCR-positive individuals and/or (2) positive for SARS CoV-2 antibody testing) were evaluated at a median time of 36 days after symptom onset (range, 18-111). Median donor age was 34.5 years (range, 20-69). Most patients had mild disease (83%) and 4 were asymptomatic, whereas 4 had moderate disease and 1 had severe disease based on World Health Organization classification, with a median of 12 days of illness (Table 1; supplemental Figure 1, available on the Blood Web site). Antibody responses were detected in 33 of the 46 convalescent donors (27/46 to spike protein and 29/46 to nucleocapsid protein; supplemental Figure 2). None of the 15 control subjects had detectable antibody responses.
Convalescent patient demographics (n = 46)
| Description | Value |
|---|---|
| Median age, y (range) | 34.5 (20-69) |
| Male | 21 (46%) |
| Disease severity | |
| Mild | 38 (83%) |
| Moderate | 3 (7%) |
| Severe | 1 (2%) |
| Asymptomatic | 4 (9%) |
| Symptoms | |
| Fever | 24 (52%) |
| Respiratory symptoms | 38 (83%) |
| Gastrointestinal symptoms | 9 (20%) |
| Fatigue | 15 (33%) |
| Anosmia | 20 (44%) |
| Median length of symptoms, d (range) | 12 (0-30) |
| Need for hospitalization | 2 (4%) |
| Description | Value |
|---|---|
| Median age, y (range) | 34.5 (20-69) |
| Male | 21 (46%) |
| Disease severity | |
| Mild | 38 (83%) |
| Moderate | 3 (7%) |
| Severe | 1 (2%) |
| Asymptomatic | 4 (9%) |
| Symptoms | |
| Fever | 24 (52%) |
| Respiratory symptoms | 38 (83%) |
| Gastrointestinal symptoms | 9 (20%) |
| Fatigue | 15 (33%) |
| Anosmia | 20 (44%) |
| Median length of symptoms, d (range) | 12 (0-30) |
| Need for hospitalization | 2 (4%) |
CSTs from convalescent donors are polyfunctional and recognize multiple viral proteins
Following stimulation and expansion of CSTs, specific T-cell activity against SARS-CoV-2 structural proteins were detected in 32 of 46 convalescent donors and 2 of 15 control subjects (Figure 1) via IFN-γ ELISpot. Convalescent donors predominantly responded to membrane (27/46, P = 6.24 × 10−6 vs control subjects), followed by spike (12/46, P = .16 vs control subjects), and nucleocapsid proteins (10/46, P = .0008 vs control subjects). Nonamplified responses to SARS-CoV-2 viral antigens were detectable from PBMCs via IFN-γ ELISpot in only 2 of 46 patients and 0 of 15 controls (supplemental Figure 3), suggesting that the frequency of the SARS-CoV-2 response is relatively low, consistent with T-cell immune responses observed against other respiratory viruses (eg, adenovirus).29,30
![T-cell recognition of SARS-CoV-2 viral antigens. Specificity of the expanded cells in response to SARS-CoV-2 antigens from convalescent patients (n = 46) and unexposed controls (n = 15) was assayed by IFN-γ ELISpot assay (bars = median). Unstimulated T cells (control [CTL] only) and stimulation with actin were used as negative controls. Results are presented as spot-forming units (SFC) per 1 × 105 cells. Specificity was defined as ≥20 spots per well with significance above background (actin) via 2-tailed Student t test. *P = .0008, **P = 6.24 × 10−6.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/25/10.1182_blood.2020008488/1/m_bloodbld2020008488f1.png?Expires=1768830659&Signature=ohlRKXS7dXrf4NSYz1mfdcRjmrG9~vdnySIyTcN7jlRAKpg4fXnLf8WdhIOJxTCSEKV30N5l5RMfBoVJYvWNBVp8agDit7koyRvrFdAnhl3~yLhy2UoFo6eNVvFrpT4Y~ondO0qywWlQJEkS~VQjOU5oH7653y3qzbtABV5~-FF1pDV8WtxD8NiyuTh~tfkyYzIwY2j7XUCoImq8J0Dk6FlwdYyKn-~LQdVjUHNcw-jL7spiz~t-mRYFnH7H6~~yWlxyX5Guva9o8Tag9b1pD9eO3VXBLfK3AnC~miCraTmJ5Wn~ecCgT1mRs8T5I3SeHP1I1ln8WxNZ81ld2Zo9YA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
T-cell recognition of SARS-CoV-2 viral antigens. Specificity of the expanded cells in response to SARS-CoV-2 antigens from convalescent patients (n = 46) and unexposed controls (n = 15) was assayed by IFN-γ ELISpot assay (bars = median). Unstimulated T cells (control [CTL] only) and stimulation with actin were used as negative controls. Results are presented as spot-forming units (SFC) per 1 × 105 cells. Specificity was defined as ≥20 spots per well with significance above background (actin) via 2-tailed Student t test. *P = .0008, **P = 6.24 × 10−6.
T-cell recognition of SARS-CoV-2 viral antigens. Specificity of the expanded cells in response to SARS-CoV-2 antigens from convalescent patients (n = 46) and unexposed controls (n = 15) was assayed by IFN-γ ELISpot assay (bars = median). Unstimulated T cells (control [CTL] only) and stimulation with actin were used as negative controls. Results are presented as spot-forming units (SFC) per 1 × 105 cells. Specificity was defined as ≥20 spots per well with significance above background (actin) via 2-tailed Student t test. *P = .0008, **P = 6.24 × 10−6.
Postexpansion T cells were predominantly CD4+, with central memory and effector memory subsets (supplemental Figure 4). The predominant CD4+ T-cell population was CXCR3+CCR6− (mean, 42.3% of CD4+ T cells) consistent with a Th1 population, with minor populations expressing CXCR5+/CXCR3− (mean, 12.95% of CD4+ T cells) and CD127−/CD25+ (mean, 15.18% of CD4+ T cells). These ratios were proportionate to rapidly expanded virus-specific T cells targeting cytomegalovirus, EBV, and adenovirus (supplemental Figure 4B). Comparatively, SARS-CoV-2-specific T cells expanded using a similar protocol in 96-well plates rather than the G-Rex10 bioreactor showed somewhat more detectable CD8 reactivity by intracellular staining (supplemental Figure 5), which may suggest that strongly elicited expansion results in preferential outgrowth of the CD4+ component.
Responses to spike and membrane proteins were confirmed to be predominantly CD4+ restricted in 11/11 tested patients (Figure 2), with significant elevations in IFN-γ/TNF-α-expressing populations targeting membrane and spike proteins (P = .008 and P = .0002 in comparison with actin, respectively). Following restimulation with viral structural proteins, CSTs produced multiple cytokines, with significant production of IL-1β, IL-2, IL-4, IL-6, IL-7, IL-12, granulocyte-macrophage colony-stimulating factor, IFN-γ, and TNF-α (supplemental Figure 6).
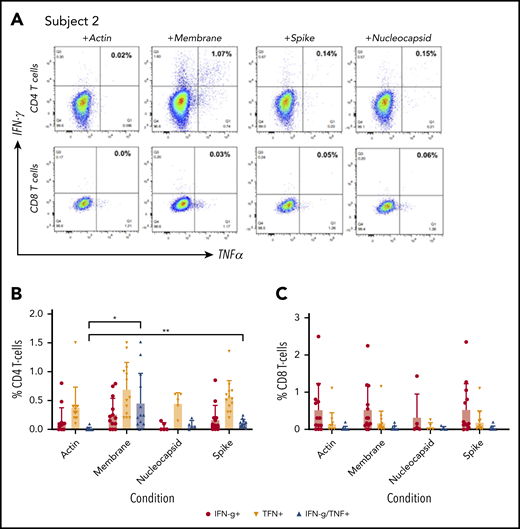
Specificity of ex vivo–expanded CST. Following 10 to 12 days of culture, specificity of CD4 and CD8 T-cell populations for membrane, spike, and nucleocapsid proteins was assessed by intracellular cytokine staining for IFN-γ and TNF-α. (A) Subject 2 demonstrates a CD4-predominant response targeting structural proteins. (B-C) Summary data of the response of expanded CD4+ T cells (B) and CD8+ T cells (C) in response to membrane, spike, and nucleocapsid proteins by intracellular cytokine staining was analyzed in convalescent donors (n = 11), and the percentage of T cells was compared with actin-stimulated controls via 2-tailed Student t test. *P < .05, **P < .01.
Specificity of ex vivo–expanded CST. Following 10 to 12 days of culture, specificity of CD4 and CD8 T-cell populations for membrane, spike, and nucleocapsid proteins was assessed by intracellular cytokine staining for IFN-γ and TNF-α. (A) Subject 2 demonstrates a CD4-predominant response targeting structural proteins. (B-C) Summary data of the response of expanded CD4+ T cells (B) and CD8+ T cells (C) in response to membrane, spike, and nucleocapsid proteins by intracellular cytokine staining was analyzed in convalescent donors (n = 11), and the percentage of T cells was compared with actin-stimulated controls via 2-tailed Student t test. *P < .05, **P < .01.
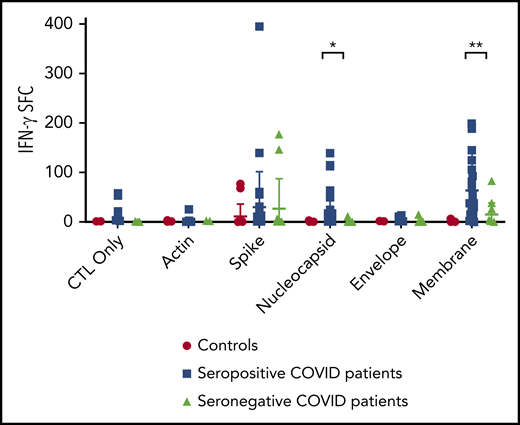
T-cell specificity of seropositive vs seronegative patients. Comparison of IFN-γ ELISpot results from postexpansion CSTs from SARS-CoV-2 seropositive vs seronegative convalescent patients was performed via Student t test. *P = .0015, **P = .00075.
T-cell specificity of seropositive vs seronegative patients. Comparison of IFN-γ ELISpot results from postexpansion CSTs from SARS-CoV-2 seropositive vs seronegative convalescent patients was performed via Student t test. *P = .0015, **P = .00075.
CSTs expanded to 18 days following a second stimulation showed a similar pattern of cytokine production, which was not statistically different from the cytokine profile following the first stimulation, with the exception of lower IFN-γ production in response to spike protein (supplemental Figure 7A). Alloreactivity testing of CSTs via 51Cr release assay showed no lysis of HLA-mismatched phytohemagglutinin blasts by T cells following up to 18 days of expansion (supplemental Figure 7B). Culture of clonal CST populations by limiting dilution and restimulation yielded several CD4+ T-cell clones, which showed polyfunctional cytokine production on peptide restimulation (supplemental Figure 8).
To assess cross-reactivity, CSTs were tested against peptides corresponding to variant epitopes in circulating SARS-CoV-2 genotypes and from the NL63 and OC43 coronaviruses.31 This testing showed moderate cross-reactivity to described variants in the regions of SARS-CoV-2 epitopes, but minimal cross-reactivity with 2 homologous nucleocapsid peptides from NL63 and OC43 (supplemental Figure 9).
CSTs from seropositive donors recognize a broader array of viral antigens than CSTs derived from donors who lack detectable humoral responses
Of the 46 convalescent patients with history of COVID-19, 26 had demonstrable antibody and T-cell responses to SARS-CoV-2. Seven convalescent donors had no detectable T-cell or antibody responses (supplemental Figure 1). Six donors had antibody responses without detectable T-cell responses and 6 donors had T-cell responses without accompanying antibody responses, as has been observed with other infections such as EBV and herpes simplex virus.32,33 A significant association was noted between presence of an antibody response and T-cell response to spike protein in convalescent patients (P = .004 via Pearson χ2 test; supplemental Figure 10). Additionally, seropositive subjects were also more likely to demonstrate a T-cell response to membrane (P = .00075) and nucleocapsid proteins (P = .0015) (Figure 3). Although there was no detectable correlation between disease severity and the magnitude of T-cell or antibody responses (supplemental Figure 11), 14 of the 20 patients who lacked T-cell and/or antibody responses had mild disease, and all 4 asymptomatic donors had incomplete immune responses (3 donors had SARS-CoV-2 T-cell responses only, and 1 donor had detectable SARS-CoV-2 antibody responses only). Evaluation of T-cell responses before COVID-19 infection was able to be performed on 2 subjects who had previously banked cells. Subject 4 had mild gastrointestinal disease, fever, and shortness of breath, and developed a CD4+ T-cell response to spike protein (which was not detectable pre-illness), but no detectable antibody response to spike or nucleocapsid. SARS-CoV-2 immune (humoral and adaptive) responses were absent in the prepandemic sample, and postinfection (after being confirmed to be PCR+ for SARS-CoV-2), a robust T-cell response to spike protein was demonstrated, though this individual did not have an antibody response to spike or nucleocapsid. Subject 46 had mild respiratory symptoms, anosmia, and gastrointestinal symptoms, and developed a T-cell response targeting spike, membrane, and nucleocapsid, as well as antibody response to both spike and nucleocapsid, both of which were absent 2 months before his illness (supplemental Figure 12).
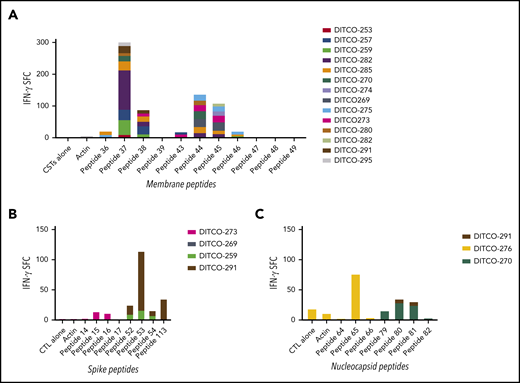
SARS-CoV-2 epitope mapping of CSTs. T-cell epitope mapping of structural proteins was performed using minipools containing 8 to 24 peptides each, with responses measured via IFN-γ ELISpot (SFC per 1 × 105 cells). (A) Epitopes within membrane protein were identified within the C terminus at AA 144-163 and 173-192, which were recognized by 8 and 6 donors, respectively. (B) Mapping of spike epitopes demonstrated three regions at AA 57-75, 205-224, and 449-463, which were recognized by 3 donors. (C) Mapping of nucleocapsid epitopes showed 2 regions at AA 357-271 and 313-335 were recognized by 3 donors.
SARS-CoV-2 epitope mapping of CSTs. T-cell epitope mapping of structural proteins was performed using minipools containing 8 to 24 peptides each, with responses measured via IFN-γ ELISpot (SFC per 1 × 105 cells). (A) Epitopes within membrane protein were identified within the C terminus at AA 144-163 and 173-192, which were recognized by 8 and 6 donors, respectively. (B) Mapping of spike epitopes demonstrated three regions at AA 57-75, 205-224, and 449-463, which were recognized by 3 donors. (C) Mapping of nucleocapsid epitopes showed 2 regions at AA 357-271 and 313-335 were recognized by 3 donors.
CSTs recognize multiple immunodominant epitopes in membrane, nucleocapsid, and spike proteins
Epitope mapping of the membrane protein yielded multiple epitopes at the C-terminal domain (Figure 4A). Two epitopes at AA 144-163 were recognized by 8 donors and were exclusively CD4-restricted (Figure 5A). Using in silico analysis, the predicted HLA restrictions of these responses were HLA-DRB1*11 and DRB4*01 (Table 2).27,28 Similarly, epitopes at AA 173-192 were recognized by 6 donors, and were also confirmed to be CD4-restricted (Figure 5B). These epitopes lie within the C-terminal domain which is located inside the virion and on intracellular membranes of infected cells that is a conserved region within all known strains of SARS-CoV2.34 Antibody blocking experiments on clonal SARS-CoV-2 CD4+ T cells demonstrated a HLA-DR restriction for several clones (supplemental Figure 13). Confirmatory restriction testing using partially HLA-matched cells confirmed that membrane peptide 37 (AA 145-160) is bound by HLA-DRB1*11:01 (supplemental Figure 14).
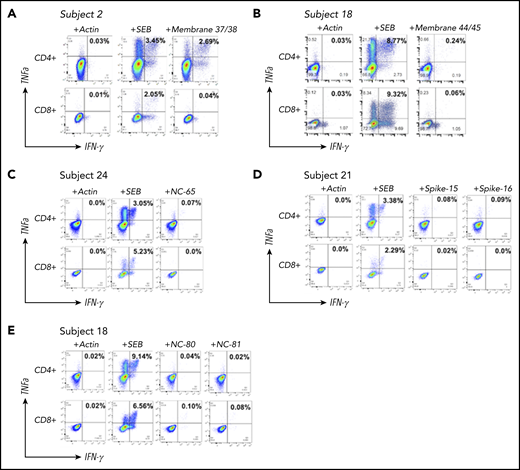
T-cell restrictions of SARS-CoV-2 epitopes. Identification of the T cells responding to each identified epitope was performed via intracellular cytokine staining on expanded CSTs, with percentages of TNF-a+/IFN-γ+ populations depicted. Intracellular cytokine staining demonstrated a predominant CD4-mediated response to membrane peptides 37-38 (A), membrane peptides 44-45 (B), nucleocapsid peptide 65 (C), and spike proteins 15-16 (D), and a predominant CD8-mediated response to nucleocapsid peptide 81 (E). SEB, staphylococcal enterotoxin β.
T-cell restrictions of SARS-CoV-2 epitopes. Identification of the T cells responding to each identified epitope was performed via intracellular cytokine staining on expanded CSTs, with percentages of TNF-a+/IFN-γ+ populations depicted. Intracellular cytokine staining demonstrated a predominant CD4-mediated response to membrane peptides 37-38 (A), membrane peptides 44-45 (B), nucleocapsid peptide 65 (C), and spike proteins 15-16 (D), and a predominant CD8-mediated response to nucleocapsid peptide 81 (E). SEB, staphylococcal enterotoxin β.
Identified class 2 epitopes in membrane, nucleocapsid, and spike proteins and predicted HLA restrictions
| Peptide sequence | Amino acid location | Subject | HLA-DRB1 | HLA-DRB3 | HLA-DRB4 | HLA-DRB5 | HLA-DQA1 | HLA-DQB1 | HLA-DPA1 | HLA-DPB1 |
|---|---|---|---|---|---|---|---|---|---|---|
| Membrane | ||||||||||
| LRGHLRIAGHHLGRC | 144-159 | 6 | 07:01, 11:04 | 02:02 | 01:03 | 02:01, 05:01 | 03:01, 03:03 | 01:03, 02:01 | 03:01, 03:03 | |
| 10 | 07:01, 11:01 | 02:02 | 01:01 | 02:01, 05:01 | 02:02, 03:01 | 01:03, 03:01 | 04:01, 11:01 | |||
| 13 | 11:04, 15:02 | 02:02 | 01:01, 01:03 | 01:02 | 01:03, 05:01 | 03:01, 06:01 | 01:03, 02:01 | 02:01, 14:01 | ||
| 18 | 11:04, 15:01 | 02:02 | 01:02, 05:01 | 03:01, 06:02 | 01:03, 02:01 | 04:01, 14:01 | ||||
| LRIAGHHLGRCDIKD | 148-163 | 6 | 07:01, 11:04 | 02:02 | 01:03 | 02:01, 05:01 | 03:01, 03:03 | 01:03, 02:01 | 03:01, 03:03 | |
| 13 | 11:04, 15:02 | 02:02 | 01:01, 01:03 | 01:02 | 01:03, 05:01 | 03:01, 06:01 | 01:03, 02:01 | 02:01, 14:01 | ||
| SRTLSYYKLGASQRV | 173-188 | 10 | 07:01, 11:01 | 02:02 | 01:01 | 02:01, 05:01 | 02:02, 03:01 | 01:03, 03:01 | 04:01, 11:01 | |
| 13 | 11:04, 15:02 | 02:02 | 01:01, 01:03 | 01:02 | 01:03, 05:01 | 03:01, 06:01 | 01:03, 02:01 | 02:01, 14:01 | ||
| 17 | 12:01, 15:01 | 02:02 | 01:01 | 01:01 | 01:02, 05:01 | 03:01, 05:01 | 01:03, 01:03 | 02:01, 04:01 | ||
| 18 | 11:04, 15:01 | 02:02 | 01:02, 05:01 | 03:01, 06:02 | 01:03, 02:01 | 04:01, 14:01 | ||||
| 21 | 03:02, 16:02 | 01:62 | 01:01, 01:03 | 02:02 | 01:02, 04:01 | 04:02, 05:02 | 02:02, 02:02 | 01:01, 01:01 | ||
| 23 | 01:01, 03:01 | 01:01 | 01:01, 01:03 | 01:01, 05:01 | 02:01, 05:01 | 01:03, 01:03 | 02:01, 02:01 | |||
| SYYKLGASQRVAGDS | 177-192 | 10 | 07:01, 11:01 | 02:02 | 01:01 | 02:01, 05:01 | 02:02, 03:01 | 01:03, 03:01 | 04:01, 11:01 | |
| 17 | 12:01, 15:01 | 02:02 | 01:01 | 01:01 | 01:02, 05:01 | 03:01, 05:01 | 01:03, 01:03 | 02:01, 04:01 | ||
| 21 | 03:02, 16:02 | 01:62 | 01:01, 01:03 | 02:02 | 01:02, 04:01 | 04:02, 05:02 | 02:02, 02:02 | 01:01, 01:01 | ||
| 23 | 01:01, 03:01 | 01:01 | 01:01, 01:03 | 01:01, 05:01 | 02:01, 05:01 | 01:03, 01:03 | 02:01, 02:01 | |||
| LGASQRVAGDSGFAA | 181-195 | 23 | 01:01, 03:01 | 01:01 | 01:01, 01:03 | 01:01, 05:01 | 02:01, 05:01 | 01:03, 01:03 | 02:01, 02:01 | |
| Nucleocapsid | ||||||||||
| KPRQKRTATKAYNVT | 257-271 | 24 | 04:01, 07:01 | 01:01 | 01:03 | 03:01, 05;05 | 03:01, 03:02 | 02:01, 02:01 | 14:01, 14:01 | |
| AFFGMSRIGMEVTPS | 313-327 | 18 | 11:04, 15:01 | 02:02 | 01:02, 05:01 | 03:01, 06:02 | 01:03, 02:01 | 04:01, 14:01 | ||
| Spike | ||||||||||
| PFFSNVTWFHAIHVS | 57-71 | 8 | 03:01, 13:01 | 01:01 | 01:03 | 01:03, 05:01 | 02:01, 06:03 | 01:03, 01:03 | 04:01, 04:01 | |
| NVTWFHAIHVSGTNG | 61-75 | 8 | 03:01, 13:01 | 01:01 | 01:03 | 01:03, 05:01 | 02:01, 06:03 | 01:03, 01:03 | 04:01, 04:01 | |
| SKHTPINLVRDLPQG | 205-219 | 37 | 03:01, 04:01 | 01:01 | 01:03 | 03:01, 05:01 | 02:01, 03:02 | 01:03, 01:03 | 02:01, 03:01 | |
| PINLVRDLPQGFSAL | 209-223 | 21 | 03:02, 16:02 | 01:62 | 01:01, 01:03 | 01:02, 04:01 | 04:02, 05:02 | 02:02, 02:02 | 01:01, 01:01 | |
| 37 | 03:01, 04:01 | 01:01 | 01:03 | 03:01, 05:01 | 02:01, 03:02 | 01:03, 01:03 | 02:01, 03:01 | |||
| YNYLYRLFRKSNLKP | 449-463 | 37 | 03:01, 04:01 | 01:01 | 01:03 | 03:01, 05:01 | 02:01, 03:02 | 01:03, 01:03 | 02:01, 03:01 |
| Peptide sequence | Amino acid location | Subject | HLA-DRB1 | HLA-DRB3 | HLA-DRB4 | HLA-DRB5 | HLA-DQA1 | HLA-DQB1 | HLA-DPA1 | HLA-DPB1 |
|---|---|---|---|---|---|---|---|---|---|---|
| Membrane | ||||||||||
| LRGHLRIAGHHLGRC | 144-159 | 6 | 07:01, 11:04 | 02:02 | 01:03 | 02:01, 05:01 | 03:01, 03:03 | 01:03, 02:01 | 03:01, 03:03 | |
| 10 | 07:01, 11:01 | 02:02 | 01:01 | 02:01, 05:01 | 02:02, 03:01 | 01:03, 03:01 | 04:01, 11:01 | |||
| 13 | 11:04, 15:02 | 02:02 | 01:01, 01:03 | 01:02 | 01:03, 05:01 | 03:01, 06:01 | 01:03, 02:01 | 02:01, 14:01 | ||
| 18 | 11:04, 15:01 | 02:02 | 01:02, 05:01 | 03:01, 06:02 | 01:03, 02:01 | 04:01, 14:01 | ||||
| LRIAGHHLGRCDIKD | 148-163 | 6 | 07:01, 11:04 | 02:02 | 01:03 | 02:01, 05:01 | 03:01, 03:03 | 01:03, 02:01 | 03:01, 03:03 | |
| 13 | 11:04, 15:02 | 02:02 | 01:01, 01:03 | 01:02 | 01:03, 05:01 | 03:01, 06:01 | 01:03, 02:01 | 02:01, 14:01 | ||
| SRTLSYYKLGASQRV | 173-188 | 10 | 07:01, 11:01 | 02:02 | 01:01 | 02:01, 05:01 | 02:02, 03:01 | 01:03, 03:01 | 04:01, 11:01 | |
| 13 | 11:04, 15:02 | 02:02 | 01:01, 01:03 | 01:02 | 01:03, 05:01 | 03:01, 06:01 | 01:03, 02:01 | 02:01, 14:01 | ||
| 17 | 12:01, 15:01 | 02:02 | 01:01 | 01:01 | 01:02, 05:01 | 03:01, 05:01 | 01:03, 01:03 | 02:01, 04:01 | ||
| 18 | 11:04, 15:01 | 02:02 | 01:02, 05:01 | 03:01, 06:02 | 01:03, 02:01 | 04:01, 14:01 | ||||
| 21 | 03:02, 16:02 | 01:62 | 01:01, 01:03 | 02:02 | 01:02, 04:01 | 04:02, 05:02 | 02:02, 02:02 | 01:01, 01:01 | ||
| 23 | 01:01, 03:01 | 01:01 | 01:01, 01:03 | 01:01, 05:01 | 02:01, 05:01 | 01:03, 01:03 | 02:01, 02:01 | |||
| SYYKLGASQRVAGDS | 177-192 | 10 | 07:01, 11:01 | 02:02 | 01:01 | 02:01, 05:01 | 02:02, 03:01 | 01:03, 03:01 | 04:01, 11:01 | |
| 17 | 12:01, 15:01 | 02:02 | 01:01 | 01:01 | 01:02, 05:01 | 03:01, 05:01 | 01:03, 01:03 | 02:01, 04:01 | ||
| 21 | 03:02, 16:02 | 01:62 | 01:01, 01:03 | 02:02 | 01:02, 04:01 | 04:02, 05:02 | 02:02, 02:02 | 01:01, 01:01 | ||
| 23 | 01:01, 03:01 | 01:01 | 01:01, 01:03 | 01:01, 05:01 | 02:01, 05:01 | 01:03, 01:03 | 02:01, 02:01 | |||
| LGASQRVAGDSGFAA | 181-195 | 23 | 01:01, 03:01 | 01:01 | 01:01, 01:03 | 01:01, 05:01 | 02:01, 05:01 | 01:03, 01:03 | 02:01, 02:01 | |
| Nucleocapsid | ||||||||||
| KPRQKRTATKAYNVT | 257-271 | 24 | 04:01, 07:01 | 01:01 | 01:03 | 03:01, 05;05 | 03:01, 03:02 | 02:01, 02:01 | 14:01, 14:01 | |
| AFFGMSRIGMEVTPS | 313-327 | 18 | 11:04, 15:01 | 02:02 | 01:02, 05:01 | 03:01, 06:02 | 01:03, 02:01 | 04:01, 14:01 | ||
| Spike | ||||||||||
| PFFSNVTWFHAIHVS | 57-71 | 8 | 03:01, 13:01 | 01:01 | 01:03 | 01:03, 05:01 | 02:01, 06:03 | 01:03, 01:03 | 04:01, 04:01 | |
| NVTWFHAIHVSGTNG | 61-75 | 8 | 03:01, 13:01 | 01:01 | 01:03 | 01:03, 05:01 | 02:01, 06:03 | 01:03, 01:03 | 04:01, 04:01 | |
| SKHTPINLVRDLPQG | 205-219 | 37 | 03:01, 04:01 | 01:01 | 01:03 | 03:01, 05:01 | 02:01, 03:02 | 01:03, 01:03 | 02:01, 03:01 | |
| PINLVRDLPQGFSAL | 209-223 | 21 | 03:02, 16:02 | 01:62 | 01:01, 01:03 | 01:02, 04:01 | 04:02, 05:02 | 02:02, 02:02 | 01:01, 01:01 | |
| 37 | 03:01, 04:01 | 01:01 | 01:03 | 03:01, 05:01 | 02:01, 03:02 | 01:03, 01:03 | 02:01, 03:01 | |||
| YNYLYRLFRKSNLKP | 449-463 | 37 | 03:01, 04:01 | 01:01 | 01:03 | 03:01, 05:01 | 02:01, 03:02 | 01:03, 01:03 | 02:01, 03:01 |
Boldface type indicates a strong binder (< 2); italic type indicates a weak binder (2-10).
Epitope mapping of spike protein yielded 3 epitopes (Figure 4B) within the S1 domain, which were also CD4-restricted (figure within the S1 domain (Figure 5D). Epitope mapping of nucleocapsid yielded CD4-restricted epitopes at AA 257-271 (Figures 4C and 5C), as well as a CD8-restricted epitope at AA 317-335 (Figure 4D; Table 3). These lie in the dimerization domain and are also highly conserved within SARS-CoV-2 genotypes (Figure 6).34,35
Identified class I epitopes in nucleocapsid and predicted HLA restrictions
| Peptide sequence | Amino acid location | Subject | HLA-A | HLA-B | HLA-C |
|---|---|---|---|---|---|
| MSRIGMEVTPSGTWL | 317-331 | 18 | 24:02, 26:01 | 40:01, 44:05* | 02:02, 03:04 |
| GMEVTPSGTWLTYTG | 321-335 | 18 | 24:02, 26:01 | 40:01, 44:05* | 02:02, 03:04 |
| Peptide sequence | Amino acid location | Subject | HLA-A | HLA-B | HLA-C |
|---|---|---|---|---|---|
| MSRIGMEVTPSGTWL | 317-331 | 18 | 24:02, 26:01 | 40:01, 44:05* | 02:02, 03:04 |
| GMEVTPSGTWLTYTG | 321-335 | 18 | 24:02, 26:01 | 40:01, 44:05* | 02:02, 03:04 |
Boldface type indicates a strong binder (<0.5); italic type indicates a weak binder (0.2-5).
Predicted B*44:05 peptide: GMEVTPSGTW.
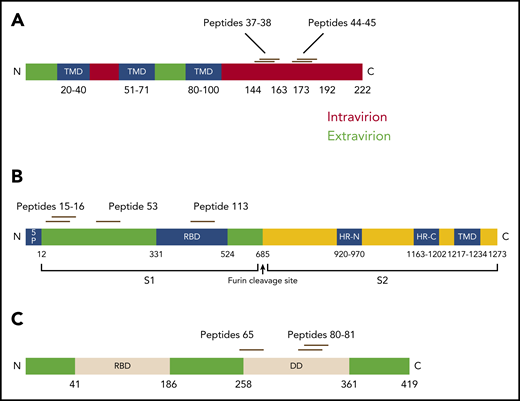
Epitope locations within SARS-CoV-2 structural proteins. (A) Epitopes within membrane protein were identified at the C-terminal intravirion domain. TMD, transmembrane domains. (B) Within spike proteins, epitopes were found within the S1 region, including 1 epitope within the receptor-binding domain (RBD). (C) In nucleocapsid protein, epitopes were identified in the region of the dimerization domain (DD).
Epitope locations within SARS-CoV-2 structural proteins. (A) Epitopes within membrane protein were identified at the C-terminal intravirion domain. TMD, transmembrane domains. (B) Within spike proteins, epitopes were found within the S1 region, including 1 epitope within the receptor-binding domain (RBD). (C) In nucleocapsid protein, epitopes were identified in the region of the dimerization domain (DD).
Discussion
Advancing knowledge of the immune response to SARS-CoV-2 is critical at the current juncture not only to guide candidate vaccine studies but, importantly, also to identify novel therapeutic targets for the design of a robust therapeutic T-cell product for the treatment of immunocompromised patients with blood disorders. Multiple studies have focused on the antibody response following COVID-19, but the persistence of antibody is unclear. Comparatively, T-cell responses are known to endure for years in response to SARS-CoV and Middle East respiratory syndrome-CoV.15,36 In immunocompromised patients, including those undergoing BMT, viruses represent a significant risk for morbidity. Though to date, relatively few immunocompromised patients have died of COVID-19 relative to the general population, prolonged illness and prolonged viral shedding has also been described, which could increase risk for other patients and staff.37,38 Furthermore, even after recovery, this population is likely to be at risk for reinfection because of compromised adaptive responses. Adoptive T-cell immunotherapy may accordingly be beneficial for prevention or early treatment of COVID-19.
In this study, we demonstrate that ex vivo–expanded CSTs may be easily generated from convalescent patients, following recovery from COVID-19, and recognize multiple immunodominant epitopes within membrane protein, which represent class II restricted T-cell epitope “hot spots.” Membrane, spike, and nucleocapsid protein showed a clear hierarchy of immunodominance, and were associated with significant increases in IFN-γ/TNF-α producing CD4+ T-cell populations. Cross-reactivity with described SARS-CoV-2 variant epitopes also suggests that T-cell responses against these regions may yield protection against circulating viral strains with these mutations. Though the understanding of the role and biologic significance of T-cell populations in combating SARS-CoV-2 remains limited, decreases in activated T-cell populations have been shown to correlate with patient acuity scores.39 Furthermore, the importance of polyfunctional CD4 T-cell responses are well-documented for antiviral immunity against other respiratory viruses.40,41 Moreover, the efficacy of adoptive, predominantly MHC class II-restricted T-cell therapies targeting adenovirus in immunocompromised patients is a prime example of the potency of T-cell therapies for clearance of respiratory viruses in the immunocompromised host.25 Though T-cell immunotherapy as treatment of infections with RNA viruses has not been attempted, the concept is supported by prior murine respiratory syncytial virus studies,42,43 and an ongoing phase 1 study has used VSTs targeting human parainfluenza-virus 3 as preventative therapy (NCT03180216). Accordingly, CSTs derived from a hematopoietic stem cell transplantation donor may be an effective preventive therapy for patients undergoing BMT. Further, for patients who lack a donor with immunity to COVID-19, the administration of partially HLA-matched third-party CSTs may be a consideration as an “on-demand” treatment of COVID-19 early in the course of infection to prevent invasive disease, with the goal to reduce the length and severity of illness.
However, the development of a potent “off-the-shelf” virus-specific T-cell therapy requires extensive characterization of the T-cell products to discover the epitope specificity and HLA restrictions of the virus specific T cells to ensure optimal matching between the virus-specific T-cell donor and the recipient. In this study, we showed that multiple regions within the highly conserved C-terminal domain of the membrane protein elicited CD4-restricted responses were shared by CST products generated from multiple individuals. The HLA restriction for membrane peptide 37 was confirmed to be mediated by HLA-DRB1*11:01, and in silico analysis suggested restriction of additional epitopes through HLA-DR11, DR7, DQ3, and DQ7, which are present in roughly 50% of the population.44 This information is therefore highly useful for the manufacture of a CST bank for clinical use. Moreover, given the increased severity of COVID-19 within minority populations, it is important to determine if there are risk associations with specific HLA types, which would need to be accounted for in candidate vaccines and understanding that these HLA restricted responses will be critical for the development of a third-party CST bank to treat the majority of screened high-risk patients (including ethnically diverse populations) as we and others have effectively achieved for other off-the-shelf virus-specific T-cell products.24,45,46 Finally, the demonstration of T-cell responses to described variant epitopes within SARS-CoV-2 suggests that CSTs are likely to have activity against many circulating viral strains in spite of genetic variation.
Overall, CSTs with specificity for ≥1 viral antigens could be successfully produced from 58% of the evaluated convalescent donors, and an association was detected between SARS-CoV-2 seropositivity and T-cell responses to non-spike antigens. It is plausible that T-follicular helper cells play a role in this association, and a population of CXCR5+ CD4+ T cells were noted in expanded CSTs. Interestingly, not all convalescent donors had detectable humoral and cellular responses, and many incongruous responses were noted. In particular, those with mild disease and those who were asymptomatic appeared to have a higher rate of seronegativity or absent T-cell responses to SARS-CoV-2 antigens, which may have implications for long-term protection for convalescent individuals, as well as for donor selection for immunotherapy. This was similarly described in several recent studies of humoral and T-cell responses.2,11 In our patients, seroconversion was noted to correlate with the presence of T-cell responses to a broader range of structural proteins. In patients who recovered from SARS-CoV disease, severity was noted to correlate with the magnitude of CD4 T-cell response,15 and it is possible a similar correlation exists in subjects with COVID-19.
Recent studies evaluating the T-cell immune response to SARS-CoV2 in unexpanded peripheral blood samples identified both CD4- and CD8-restricted responses to viral structural proteins in convalescent donors, as well as in a fraction of unexposed subjects.4-11 Prior studies have postulated that this may be due to cross-reactivity with common circulating coronaviruses. In our study, we also observed T-cell responses to spike proteins in 2 of 15 unexposed control subjects. However, we did not observe any responses to nucleocapsid or membrane proteins, which also paralleled our observation that responses to these proteins were predominantly detected in subjects with confirmed humoral immunity (ie, seropositivity). Although we cannot definitively rule out rare T-cell populations recognizing nonspike proteins in virus-naïve donors, the absence of these responses in our study, even following ex vivo expansion, suggests that T-cell reactivity in unexposed individuals is more limited than in seropositive convalescent patients, which may reflect the differences in structural proteins in SARS-CoV-2 vs other commonly circulating coronaviruses (Table 4). Larger, longitudinal studies to analyze the cellular response to other coronaviruses and their possible cross-reactivities with SARS-CoV-2 will therefore be necessary to understand the clinical implications of preexisting T-cell responses to SARS-CoV-2 antigens. Whether T-cell responses in unexposed donors may be effectively harnessed through selection, rapid expansion, or through methods akin to generation of CMV-specific T-cells from naïve donors, will also require study.47,48 Nevertheless, given the information currently available, our recommendation would be that seropositivity may not be necessary for the generation of a donor-derived CST product to be given prophylactically in the BMT setting. In contrast, for the development of a third party off-the-shelf CST therapeutic for the treatment of high-risk patients with known infection, our data suggests that using donors with confirmed humoral immunity will enable the generation of broadly antigen and epitope specific therapeutic T-cell products.
Epitope homology with other human coronaviruses
| SARS-CoV-2 epitope identified | Other human coronavirus | Protein name | Amino acid sequence alignment |
|---|---|---|---|
| Membrane | |||
| LRGHLRIAGHHLGRC | LRGHLRIAGHHLGRC | ||
| SARS coronavirus | M protein | RGHLRMAGHPLGRC | |
| HKU1 | Membrane glycoprotein | RGHLYIQGVKLG | |
| MERS | M protein | GHLKIAGMHFGAC | |
| LRIAGHHLGRCDIKD | LRIAGHHLGRCDIKD | ||
| SARS coronavirus | Membrane protein | LRMAGHPLGRCDIKD | |
| SRTLSYYKLGASQRV | SRTLSYYKLGASQRV | ||
| SARS coronavirus | Membrane protein | SRTLSYYKLGASQRV | |
| SYYKLGASQRVAGDS | SYYKLGASQRVAGDS | ||
| SARS coronavirus | Membrane protein | SYYKLGASQRVGTDS | |
| LGASQRVAGDSGFAA | LGASQRVAGDSGFAA | ||
| SARS Coronavirus | Membrane glycoprotein | LGASQRVGTDSGFAA | |
| NL63 | Orf1a protein | LGAS–VTEDVKFAA | |
| Nucleocapsid | |||
| KPRQKRTATKAYNVT | KPRQKRTATKAYNVT | ||
| SARS coronavirus | Nucleocapsid protein | KPRQKRTATKQYNVT | |
| OC43 | Nucleocapsid protein | KPRQKRSPNK | |
| NL63 | Chain A, nucleocapsid | KPRWKRVPTREENV | |
| MERS | Nucleocapsid protein | RHKRVATKSFNV | |
| AFFGMSRIGMEVTPS | AFFGMSRIGMEVTPS | ||
| SARS coronavirus | Nucleocapsid protein N | AFFGMSRIGMEVTPS | |
| MSRIGMEVTPSGTWL | MSRIGMEVTPSGTWL | ||
| SARS coronavirus | Nucleocapsid protein | MSRIGMEVTPSGTWL | |
| GMEVTPSGTWLTYTG | GMEVTPSGTWLTYTG | ||
| SARS coronavirus | Nucleocapsid protein | GMEVTPSGTWLTY | |
| Spike | |||
| PFFSNVTWFHAIHVS | — | — | — |
| NVTWFHAIHVSGTNG | — | — | — |
| SKHTPINLVRDLPQG | SKHTPINLVRDLPQG | ||
| OC43 | Replicase polyprotein 1ab | PANIV—LPQG | |
| SARS coronavirus | S1 protein | PIDVVRDLPSG | |
| PINLVRDLPQGFSAL | PINLVRDLPQGFSAL | ||
| SARS coronavirus | Chain A, spike | PIDVVRDLPSGFNTL | |
| OC43 | Replicase polyprotein 1ab | PANIV–LPQG | |
| YNYLYRLFRKSNLKP | YNYLYRLFRKSNLKP | ||
| SARS coronavirus | Chain E, spike glycoprotein | YNYKYRYLRHGKLRP |
| SARS-CoV-2 epitope identified | Other human coronavirus | Protein name | Amino acid sequence alignment |
|---|---|---|---|
| Membrane | |||
| LRGHLRIAGHHLGRC | LRGHLRIAGHHLGRC | ||
| SARS coronavirus | M protein | RGHLRMAGHPLGRC | |
| HKU1 | Membrane glycoprotein | RGHLYIQGVKLG | |
| MERS | M protein | GHLKIAGMHFGAC | |
| LRIAGHHLGRCDIKD | LRIAGHHLGRCDIKD | ||
| SARS coronavirus | Membrane protein | LRMAGHPLGRCDIKD | |
| SRTLSYYKLGASQRV | SRTLSYYKLGASQRV | ||
| SARS coronavirus | Membrane protein | SRTLSYYKLGASQRV | |
| SYYKLGASQRVAGDS | SYYKLGASQRVAGDS | ||
| SARS coronavirus | Membrane protein | SYYKLGASQRVGTDS | |
| LGASQRVAGDSGFAA | LGASQRVAGDSGFAA | ||
| SARS Coronavirus | Membrane glycoprotein | LGASQRVGTDSGFAA | |
| NL63 | Orf1a protein | LGAS–VTEDVKFAA | |
| Nucleocapsid | |||
| KPRQKRTATKAYNVT | KPRQKRTATKAYNVT | ||
| SARS coronavirus | Nucleocapsid protein | KPRQKRTATKQYNVT | |
| OC43 | Nucleocapsid protein | KPRQKRSPNK | |
| NL63 | Chain A, nucleocapsid | KPRWKRVPTREENV | |
| MERS | Nucleocapsid protein | RHKRVATKSFNV | |
| AFFGMSRIGMEVTPS | AFFGMSRIGMEVTPS | ||
| SARS coronavirus | Nucleocapsid protein N | AFFGMSRIGMEVTPS | |
| MSRIGMEVTPSGTWL | MSRIGMEVTPSGTWL | ||
| SARS coronavirus | Nucleocapsid protein | MSRIGMEVTPSGTWL | |
| GMEVTPSGTWLTYTG | GMEVTPSGTWLTYTG | ||
| SARS coronavirus | Nucleocapsid protein | GMEVTPSGTWLTY | |
| Spike | |||
| PFFSNVTWFHAIHVS | — | — | — |
| NVTWFHAIHVSGTNG | — | — | — |
| SKHTPINLVRDLPQG | SKHTPINLVRDLPQG | ||
| OC43 | Replicase polyprotein 1ab | PANIV—LPQG | |
| SARS coronavirus | S1 protein | PIDVVRDLPSG | |
| PINLVRDLPQGFSAL | PINLVRDLPQGFSAL | ||
| SARS coronavirus | Chain A, spike | PIDVVRDLPSGFNTL | |
| OC43 | Replicase polyprotein 1ab | PANIV–LPQG | |
| YNYLYRLFRKSNLKP | YNYLYRLFRKSNLKP | ||
| SARS coronavirus | Chain E, spike glycoprotein | YNYKYRYLRHGKLRP |
Boldface type indicates a strong binder (< 2).
MERS, Middle East respiratory syndrome.
Limitations of this study include the sample size, the tendency toward mild illness in the subjects, and that not all the subjects were PCR-positive or antibody-positive for SARS-CoV-2. However, because the vast majority of the convalescent donors had uncomplicated disease, our data suggest that T-cell and humoral responses measured here represent an effective adaptive immune response to SARS-CoV-2 that can be effectively harnessed (especially from BMT donors) for the manufacture of CST products for clinical use. We did not evaluate donors longitudinally, and therefore the absence of T-cell responses in 42% of subjects may relate to the timing of T-cell responses following primary infection. We limited evaluation to structural viral proteins, given their described immunodominance in related coronaviruses, but it is possible that T-cell responses to nonstructural proteins may have been present, as has been demonstrated in recent studies. The study was also inadequately powered to determine if any correlations exist between clinical severity and recognition of specific T-cell epitopes. However, as all of the evaluated patients survived and recovered without significant inflammatory or thrombotic complications, it is a reasonable assumption that the detected T-cell responses represent beneficial adaptive cellular responses.
We do acknowledge that a maladaptive immune response is highly suspected to be the cause of hyperinflammatory complications such as multisystemic inflammatory syndrome in children,49 and an understanding of the role of adaptive and innate responses in patients with inflammatory complications will be critical in determining the characteristics of an effective and enduring adaptive immune response to SARS CoV-2. Although this is a consideration when developing adoptive T-cell immunotherapy trials for this disease, the detection of T cells in our cohort of donors, the majority of whom had mild disease suggests that judicious use of CST products, given early in the infection process or given as prophylaxis (eg, early post-BMT) to high-risk patients warrants investigation. Inflammatory complications in patients with COVID-19 have been correlated with elevation of IL-6, IL-10, and IL-13.50,51 Because other inflammatory complications such as cytokine release syndrome are very rare after virus-specific T-cell therapy,52 the risk of inflammatory complications after adoptive T-cell therapy for COVID-19, particularly when used early and derived from donors who themselves did not have inflammatory disease, are likely low.
In summary, this is the first report to demonstrate that a broadly specific T-cell therapeutic targeting 3 structural proteins of SARS-CoV-2 can be reliably expanded using GMP-compliant methodologies from the majority of convalescent donors. Moreover, the CST products are principally comprised CD4+ T cells specific for conserved regions of these proteins, and most frequently, the membrane protein. The immunodominance of the membrane protein therefore also has important implications for vaccine development to elicit cellular immune responses because most current vaccine candidates are focused exclusively on the spike protein to elicit neutralizing antibody. This work now enables the rapid translation of this novel treatment to the clinic. Future studies will therefore evaluate whether patient-specific and/or off-the-shelf adoptive T-cell immunotherapies using this novel CST product will emerge as a safe and useful treatment modality in high-risk patients with COVID-19, as we and others have effectively shown for the treatment of other respiratory viruses especially in the BMT setting.53
For original data, please e-mail the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the staff members of the Center for Cancer and Immunology Research and the National Institute of Allergy and Infectious Diseases (NIAID) Laboratory of Infectious Disease, and our referring collaborators for enabling this work. The authors also are very grateful to all their patients and their families for participating in this work.
This work was supported by grants from the National Institutes of Health (NIH) National Heart, Lung, and Blood Institute (K23-HL136783-01) (M.D.K.), the Jeffrey Modell Foundation, the Board of Visitors of the Children’s National Health System, the Katzen Foundation, the Connor Family Foundation, and the intramural research programs of the NIH/NIAID and the NIH/National Institute of Dental and Craniofacial Research. This work was also supported, in part, by a COVID-19 Research Grant to J.M.B. funded by the Weill Cornell Medicine Board of Overseers and additional donors.
Authorship
Contribution: M.D.K., C.A.L., A.A.A., P.J.H., C.R.C., R.B.J., J.I.C., and C.M.B. conceived and designed the experiments; K.M.H., M.A.J.-W., V.V.K., J.D.-S., P.-H.L., K.C., K.W., A.D., M.T., E.K.R., E.M.S., S.V., Z.S., N.Z., U.E., M.S., A.G., R.U., F.H., K.F., L.D., K.G., P.D.B., and R.B.J. conducted the research; M.D.K., K.M.H., C.A.L., P.-H.L., H. Liang, J.I.C., and C.M.B. analyzed data; M.D.K., K.M.H., A.A.A., P.J.H., C.R.C., R.B.J., J.I.C., and C.M.B. wrote the manuscript; and all authors have read and approved the final manuscript.
Conflict-of-interest disclosure: P.J.H. and C.R.C. are cofounders and are on the board of directors or scientific advisory board of Mana Therapeutics. P.J.H. is on a scientific advisory board for Cellevolve. C.R.C. is a consultant for Catamaran Bio. C.M.B. is on the advisory board for Cellectis and is cofounder and on the scientific advisory boards for Catamaran Bio and Mana Therapeutics with stock and/or ownership, is on the board of directors for Caballeta Bio with stock options and has stock in Neximmune and Torque Therapeutics. M.D.K. is on a scientific advisory panel for Gilead Sciences. K.M.H., M.D.K., C.A.L., P.J.H., C.R.C., A.A.A., and C.M.B. have filed a patent application based on the findings in this paper. The remaining authors declare no competing financial interests.
Correspondence: Catherine M. Bollard, Center for Cancer and Immunology Research, Children’s National Health System, 111 Michigan Ave, NW, Washington DC 20010; e-mail: cbollard@childrensnational.org.
