

Key Points
siRNA targeting FXIII-B decreases the concentration of plasma FXIII-A for more than 3 weeks after a single injection.
Pharmacologic depletion of FXIII-A via FXIII-B can enhance fibrinolysis without excessive bleeding.

Abstract
The activated form of coagulation factor XIII (FXIII-A2B2), FXIII-A*, is a hemostatic enzyme essential for inhibiting fibrinolysis by irreversibly crosslinking fibrin and antifibrinolytic proteins. Despite its importance, there are no modulatory therapeutics. Guided by the observation that humans deficient in FXIII-B have reduced FXIII-A without severe bleeding, we hypothesized that a suitable small interfering RNA (siRNA) targeting hepatic FXIII-B could safely decrease FXIII-A. Here we show that knockdown of FXIII-B with siRNA in mice and rabbits using lipid nanoparticles resulted in a sustained and controlled decrease in FXIII-A. The concentration of FXIII-A in plasma was reduced by 90% for weeks after a single injection and for more than 5 months with repeated injections, whereas the concentration of FXIII-A in platelets was unchanged. Ex vivo, crosslinking of α2-antiplasmin and fibrin was impaired and fibrinolysis was enhanced. In vivo, reperfusion of carotid artery thrombotic occlusion was also enhanced. Re-bleeding events were increased after challenge, but blood loss was not significantly increased. This approach, which mimics congenital FXIII-B deficiency, provides a potential pharmacologic and experimental tool to modulate FXIII-A2B2 activity.
Introduction
Transglutaminases are a major class of enzymes that catalyze the posttranslational formation of isopeptide bonds within and between proteins. Although several transglutaminases have been implicated in diseases, specific therapeutic inhibitors have been challenging to develop because of their high structural homology.1 The most abundant transglutaminase in blood is coagulation factor XIII (FXIII). FXIII is present in plasma as a heterotetrameric proenzyme (FXIII-A2B2), where it is proteolyzed by thrombin in the presence of Ca2+ to become the activated enzyme, FXIII-A*. Its best understood function is to facilitate fibrin-fibrin and fibrin-antifibrinolytic protein crosslinks, thereby stabilizing the clot and inhibiting fibrinolysis. The FXIII-B subunit functions to stabilize FXIII-A in the blood, which extends the circulating half-life of FXIII-A from 3 days to 11 days.2-5 In addition to the reservoir of FXIII-A2B2 in the blood, the homodimeric proenzyme FXIII-A2 is expressed in platelets, megakaryocytes, monocytes, macrophages, and osteoblasts. In concert with plasma FXIII-A2B2, cellular FXIII-A2 contributes to hemostasis, wound healing, phagocytosis, and bone and matrix remodeling.6,7
Plasma-derived FXIII-A2B2 has been linked to pathologies, including venous thrombosis,8 cerebral amyloid angiopathy,9 myocardial infarction,10 arthritis,11 and cancer metastasis.12 A pharmacologic inhibitor specific for FXIII-A2B2 or FXIII-A* that is effective in vivo is needed. FXIII-A2B2 is considered a pharmacologic target for preventing venous thrombosis because FXIII increases clot stability, retention of red blood cells, and thrombus weight.13,14 Thrombosis caused by coronavirus disease 2019 (COVID-19)–associated coagulopathy and the limitations of direct oral anticoagulants and heparin in COVID-19–associated coagulopathy highlight the need for novel approaches to antithrombotic therapy.15,16 Although some COVID-19 patients have benefited from thrombolytic therapy,17 that therapy is associated with severe bleeding.18 An agent (potentially in combination with other agents) that enhances endogenous fibrinolysis is an alternative approach to safely preventing and alleviating thrombosis.
Despite the demand for inhibitors of FXIII,19-22 the current approaches to inhibition are limited.23 Mice with the F13A gene deleted have provided insight into the role of the FXIII-A subunit in vivo.14 Generating gene-targeted animals is costly and time consuming, making analysis of FXIII in non-murine species or mice with additional gene knockouts challenging. The available small molecule enzymatic inhibitors of FXIII-A* are limited by low specificity or poor pharmacokinetics in vivo that render them ineffective for studies that last more than a few hours.21,24 As in F13A gene–deleted animals, enzymatic inhibitors cannot differentiate between the activity of plasma-derived and surface-exposed cellular FXIII-A*,25 so another technology must be developed.
FXIII deficiency in humans is a rare bleeding disorder that provides insight into approaches to developing modulatory therapeutics. Patients can exhibit congenital or acquired deficiency of either the FXIII-A or -B subunit or dysfunctional FXIII-A*.26-28 In all cases, there is decreased activity of plasma FXIII-A*, but the bleeding phenotype varies considerably depending on the amount of residual FXIII-A activity.26,27,29 Severe congenital FXIII-A deficiency via homozygous or compound heterozygous mutations in the F13A1 gene causes both severe bleeding and impaired wound healing because of a lack of both plasma and cellular FXIII-A.14,28,30 In contrast, deficiency in FXIII-B leads to depletion of only plasma FXIII-A31 and is generally not associated with severe bleeding.26,32,33 The phenotypes of patients with FXIII-B deficiency suggest that pharmacologic depletion of FXIII-B would safely and specifically decrease the concentration of plasma FXIII-A2B2, which would result in reduced FXIII-A* activity in vivo and enhanced fibrinolysis.
The cellular synthesis of FXIII-B is an ideal target for small interfering RNA (siRNA). When packaged in lipid nanoparticles (LNPs) containing ionizable cationic lipids, appropriate siRNA sequences can efficiently modulate the expression of proteins produced in the liver and released into plasma.34 Ionizable cationic LNPs undergo uptake into hepatocytes via the low-density lipoprotein receptor, facilitated by the binding of apolipoprotein E;35 siRNA facilitates the degradation of its target messenger RNA (mRNA) in hepatocytes within hours of intravenous (IV) administration. The reduction of mRNA leads to a decrease in the target protein concentration according to the protein half-life. This has been an effective method for knockdown of transthyretin,36 antithrombin,37 FXI,38 FXII,39 protein C,40 and fibrinogen.41 FXIII-B is synthesized nearly exclusively in the liver, whereas FXIII-A is synthesized in many tissues that are not accessible to current siRNA approaches such as the bone marrow. Formulations of LNPs with specific ionizable cationic lipids and siRNA sequences have been safe and efficacious in patients, with repeat dosing every 3 weeks for more than 18 months.42 In this study, we tested the hypothesis that siRNA targeting FXIII-B (siFXIIIB) reduces plasma FXIII-A concentration and activity and enables long-acting prophylactic enhancement of fibrinolysis in mice.
Materials and methods
siRNA-LNP preparation, analysis, and administration
2′O-methylated siRNA, obtained commercially (Integrated DNA Technologies, Coralville, IA) was dissolved in 25 mM sodium acetate (pH 4) buffer at an amine:phosphate ratio of 3. DLin-MC3-DMA, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), cholesterol, and dimyristoyl glycerol-polyethyleneglycol (DMG-PEG) lipids were dissolved in ethanol at a molar ratio of 50:10:38.5:1.5 mol%, respectively, to achieve a final concentration of 20 mM total lipid. The 2 solutions were mixed using a T-junction mixer as described previously.43 The resulting lipid nanoparticles were dialyzed against phosphate-buffered saline (pH 7.4) in a 1000-fold excess. Cholesterol content was measured by using a Cholesterol E Assay Kit (Wako Chemicals, Mountain View, CA), from which total lipid concentration was extrapolated. Nucleic acid entrapment was determined using a RiboGreen assay.44 siRNA-LNPs were diluted to a concentration of 0.1 mg of siRNA per mL and administered to mice at a dose of 1 mg siRNA per kg of body weight via tail vein injection. siRNA targeting FXIII-B (siFXIIIB) was used as treatment, and siRNA targeting luciferase (siLuc) was used as a control. Ldlr−/− mice were injected retro-orbitally with siRNA-LNPs.
Animal blood draws and platelet isolation
Murine studies were performed in accordance with protocols approved by the University of British Columbia and University of Cincinnati Animal Care Committees. C57BL/6J (stock #000664; The Jackson Laboratory, Bar Harbor, ME) and B6.129S7-Ldlrtm1Her/J (Ldlr−/−; originating at The Jackson Laboratory and bred in Owens Laboratory) mice were used in all studies. Blood samples used to assess plasma protein levels were collected via saphenous vein puncture (C57BL/6J mice) into heparinized capillaries or the inferior vena cava (Ldlr−/− mice) via a 25G needle. Blood was drawn for coagulation assays from mice anesthetized with isoflurane by cardiac puncture using a 23G needle containing sodium citrate (109 mM) to a final v/v concentration of 10% in whole blood. To collect plasma and platelets, whole blood was first spun at 600g for 10 minutes, and the supernatant including the intermediate platelet layer was collected. Subsequently, platelet rich plasma (PRP) was spun at 800g for 10 minutes. The resulting platelet pellet was resuspended and washed using Tyrode’s buffer (pH 6.5).
Whole blood was collected from rabbits via ear vein puncture using a 22G needle containing sodium citrate (109 mM) to a final v/v concentration of 10% in whole blood. To collect plasma, whole blood was first spun at 160g for 10 minutes, and the supernatant including the intermediate platelet layer was collected. Subsequently, PRP was spun twice at 400g for 15 minutes.
mRNA quantification
Livers were surgically removed from anesthetized mice, and tissue was homogenized in Trizol (Thermo Fisher, Waltham, MA). Nucleic acid was extracted by phenol-chloroform precipitation. DNA was digested by incubating the sample with TURBO DNase (Thermo Fisher) at 37°C for 1 hour. DNase was removed by repeating the Trizol-chloroform extraction. Reverse transcription was performed using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA) followed by quantitative polymerase chain reaction with SYBR Green Master Mix (Thermo Fisher) and DNA primers (Integrated DNA Technologies) against FXIII-B (forward: CAAACGGTGTGGTTGTTGATG; reverse: CAAGCAGACAGGTGGAGATGAC and Ppia (forward: GCGTCTCCTTCGAGCTGTT; reverse: TGTAAAGTCACCACCCTGGC).
Western blotting
Samples were reduced, boiled, and separated on 4% to 15% acrylamide gradient gels (Bio-Rad). After electrophoresis, the samples were transferred to a nitrocellulose membrane (GE Healthcare, Chicago, IL) and blocked with Odyssey Blocking Buffer (LI-COR, Lincoln, NE). The membranes were treated with a primary antibody against FXIII-A (1:1000; SAF13A-AP; confirmed cross-reactivity: human, rat, mouse, rabbit, canine; Affinity Biologicals, Ancaster, ON, Canada), platelet factor 4 (1:1000; SAPF4-AP; confirmed cross-reactivity: not determined; both from Affinity Biologicals), or fibrin (1:1000; 4440-8004; confirmed cross-reactivity: mouse, rat; Bio-Rad), washed, and treated with horseradish peroxidase–labeled anti-host secondary antibody (1:15 000; Abcam, Cambridge, MA). Specific bands were imaged using Clarity ECL (Bio-Rad) on film (Mandel, Guelph, ON, Canada). Quantification of western blots was done using ImageJ image processing software (National Institutes of Health, Bethesda, MD) to measure band intensity relative to background and loading controls.
Thromboelastography (TEG)
Shear elastic moduli were evaluated at 37°C using a TEG Hemostasis Analyzer System 5000 (Haemoscope Corp., Niles, IL). Citrated mouse whole blood or rabbit plasma was combined with CaCl2 (10 mM), thrombin (0.03 nM Innovin; MedCorp, São Paulo, Brazil), and tissue plasminogen activator (tPA; 3.8 nM) over 3 hours.
Administration of purified murine FXIII-A2B2 (mFXIII)
mFXIII (Enzyme Research Laboratories, Uplands, United Kingdom) was sterile filtered, and diluted in phosphate-buffered saline to a concentration of 0.068 mg/mL. mFXIII (10 µL/g body weight) was administered via tail vein injection into 2 mice previously treated with siFXIIIB at a dose of 0.68 µg/g body weight (∼12 µg/mL blood or 40% of normal concentration of FXIII-A2B2).
Fibrin crosslinking in plasma
Plasma from mice 14 days after administration or rabbits 42 days after administration was incubated with bovine thrombin (70 nM) with or without CaCl2 (4 mM), T101 (0.8 mM), an inhibitor of FXIII-A* (Zedira, Darmstadt, Germany), or an inhibitor specific for tissue transglutaminase Z006 (Z-DON-Val-Pro-Leu-OMe; 0.8 mM; Zedira) at 37°C. The samples were treated with reaction-quenching buffer (8 M urea, 50 mM dithiothreitol, 12.5 mM EDTA) for at least 1 hour at 60°C to solubilize the clot. Clot lysates were analyzed by western blot as described above.
FeCl3 model of arterial thrombosis
The carotid arteries of C57BL/6J mice (pretreated with siLuc or siFXIIIB) were exposed, and a 0.5 mm Transonic Doppler ultrasound flow probe was attached (ADInstruments, Colorado Springs, CO) as described.45 Flow rate data were acquired using Powerlab and LabChart software (ADInstruments). A piece of filter paper (1 × 1 mm) soaked in 10% (w/v) FeCl3 was applied to the vessel for 3 minutes followed by a saline rinse. Once vessel occlusion was stable, as seen by the Doppler ultrasound, tenecteplase at a dose of 9 mg/kg was administered intravenously via the tail vein. To quantify reperfusion in the following observation period, the total blood flow was calculated from the area of the Doppler ultrasound curve (volt/s). Because blood flow could vary between mice before occlusion, the total blood flow after occlusion was corrected using the pre-occlusion flow rate in each mouse, as previously described.45
Tail transection bleeding model
Apixaban (Eliquis; Bristol-Meyer Squibb, New York, NY) 5-mg tablets were crushed and dissolved to a concentration of 0.204 mg/mL. Apixaban solution was administered to mice via oral gavage at a final dose of 1 mg/kg. This dose is equivalent to a standard therapeutic dose (0.083 mg/kg) converted to the equivalent murine dose using body surface area ratios from the US Food and Drug Administration guidelines.46 Two hours after apixaban administration or 2 weeks after siFXIIIB administration, C57BL/6J mice were anesthetized with 10% to 15% isoflurane and were kept on a heating pad to maintain an internal temperature of 36 ± 2°C. The tail was transected at 2.5 mm diameter and immersed in saline at 37°C. The severity of bleeding was recorded every 60 seconds over 40 minutes; the severity was scored between 0 and 5 (0 indicated no bleeding and 5 indicated a consistent stream of blood loss of approximately 35 µL/minute). The number of spontaneous re-bleeds were counted, blood from the tail bleed was collected, and hemoglobin concentration was measured via spectrophotometry.47
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8.0.1. For groups of sufficient size (n ≥ 3), significance was assessed. Two-tailed unpaired Student t test was used to compare 2 data sets. Two-way analysis of variance (ANOVA) was used to compare 2 data sets over time, and 1-way ANOVA was used to compare multiple data sets with 1 variable. Welch’s ANOVA was used when variance was not equal (Brown-Forsythe), and all data had normal distribution (Shapiro-Wilk). Significance was designated at P < .05.
Results
siFXIIIB depleted plasma FXIII-A without affecting platelet FXIII-A
A specific siRNA sequence was designed to knock down FXIII-B mRNA in mice (siFXIIIB). siFXIIIB was encapsulated in LNPs containing an ionizable cationic lipid and administered to mice with a single IV injection at 1 mg siRNA per kg body weight. At weekly intervals, FXIII-B mRNA was measured in surgically excised liver tissue using quantitative polymerase chain reaction and was compared with Ppia as a housekeeping control in mouse hepatocytes.48 Within 24 hours, mRNA encoding FXIII-B decreased by 89% ± 1% compared with that in untreated mice, whereas FXIII-B mRNA in control mice treated with siRNA against luciferase (siLuc) remained unchanged (Figure 1A; supplemental Figure 1, available on the Blood Web site). The concentration of plasma FXIII-A protein was reduced by 97% ± 2% within 11 days of the siFXIIIB injection compared with siLuc mice (Figure 1B-C). The knockdown of FXIII-B mRNA persisted for over 3 weeks (P < .05 during week 2 and P < .01 during week 3) and induced a corresponding decrease in FXIII-A protein concentration (P < .01 during weeks 2 and 3). mRNA and protein levels began to return to baseline in the fourth week. Throughout the 28 days, FXIII-A concentrations remained unchanged in washed platelets from the same blood samples (Figure 1D). This demonstrates that siFXIIIB effectively knocks down only the plasma fraction of FXIII-A. To define the potential of siFXIIIB to sustain the knockdown of plasma FXIII-A, we treated a cohort of mice once every 3 weeks for 150 days. This regimen maintained the knockdown at concentrations between 1% and 19% of the pretreatment levels for more than 5 months (Figure 1E). No significant difference in FXIII-A was observed in Ldlr−/− mice 2 weeks after treatment with LNPs containing siLuc or siFXIIIB (supplemental Figure 2), consistent with the known mechanism of transfection of LNPs in hepatocytes.

siFXIIIB decreased FXIII-B mRNA in the livers of mice, leading to a sustained decrease of FXIII-A protein in plasma but not platelets for more than 3 weeks. Mice were injected with a single dose of siFXIIIB (green) or siLuc (lavender). (A) mRNA encoding FXIII-B was measured in liver tissue using quantitative polymerase chain reaction normalized against a housekeeping gene Ppia and graphed relative to FXIII-B mRNA from untreated mice. (B) Representative western blots against FXIII-A, in which each lane contains the plasma from an individual mouse in either treatment group. The triangular marker indicates the expected molecular weight of FXIII-A (83 kDa). (C) Quantification of panel B using densitometry, normalized to a loading control, and graphed relative to FXIII-A antigen from untreated control mice; n = 3 mice per time point. (D) Representative western blot against FXIII-A in PPP and washed platelets from mice treated with siFXIIIB 14 days previously. The quantification is normalized to loading controls platelet factor 4 (platelets) or immunoglobulin G (plasma) and relative to untreated control. (E) Plasma FXIII-A quantified at various time points after repeated injections of siFXIIIB relative to starting concentrations before treatment; n = 3 mice per time point; vertical gray dashed lines indicate times of injections. For all graphs, values represent mean ± standard error of the mean (SEM). ns, not significant (P > .05); *P < .05; **P < .01.
siFXIIIB decreased FXIII-B mRNA in the livers of mice, leading to a sustained decrease of FXIII-A protein in plasma but not platelets for more than 3 weeks. Mice were injected with a single dose of siFXIIIB (green) or siLuc (lavender). (A) mRNA encoding FXIII-B was measured in liver tissue using quantitative polymerase chain reaction normalized against a housekeeping gene Ppia and graphed relative to FXIII-B mRNA from untreated mice. (B) Representative western blots against FXIII-A, in which each lane contains the plasma from an individual mouse in either treatment group. The triangular marker indicates the expected molecular weight of FXIII-A (83 kDa). (C) Quantification of panel B using densitometry, normalized to a loading control, and graphed relative to FXIII-A antigen from untreated control mice; n = 3 mice per time point. (D) Representative western blot against FXIII-A in PPP and washed platelets from mice treated with siFXIIIB 14 days previously. The quantification is normalized to loading controls platelet factor 4 (platelets) or immunoglobulin G (plasma) and relative to untreated control. (E) Plasma FXIII-A quantified at various time points after repeated injections of siFXIIIB relative to starting concentrations before treatment; n = 3 mice per time point; vertical gray dashed lines indicate times of injections. For all graphs, values represent mean ± standard error of the mean (SEM). ns, not significant (P > .05); *P < .05; **P < .01.
FXIII-B knockdown increased clot susceptibility to fibrinolysis by delaying crosslinking of proteins
TEG was used to measure the stability of clots formed ex vivo in blood collected from mice treated with siFXIIIB or siLuc. Maximal knockdown was achieved 2 weeks after administration of the siFXIIIB, so mice were evaluated at this time point in subsequent experiments. R time and maximum amplitude were not significantly different between the 2 groups; however, siFXIIIB led to reduced time to fibrinolysis. When tPA was added to the samples before clotting, blood from mice treated with siFXIIIB had significantly more lysis at 30 minutes (P = .004) and a 1.8-fold shorter total lysis time (P = .004) (Figure 2A-E). To reverse the knockdown, purified mFXIII was administered intravenously to 2 mice at a dose of 0.68 µg per g of body weight (∼12 µg/mL blood or 40% of normal concentration of FXIII). TEG indicated that with this dose of purified FXIII, clot resistance to fibrinolysis was partially recovered (Figure 2F-G). Mice pretreated with siFXIIIB had, on average, 2.8-fold faster total lysis time than untreated mice, and this was decreased to 1.4-fold by IV exogenous FXIII.

siFXIIIB delays crosslinking of α2-antiplasmin, leading to clots that are more susceptible to lysis ex vivo. (A-B) Representative TEG curve tracings using blood from mice treated with siLuc (A, lavender) or with siFXIIIB (B, green). In each case, recombinant tissue factor was added to stimulate clotting and 4 nM of tPA was added to enhance lysis. (C-E) TEG curves were quantified for (C) maximum clot stiffness, (D) percent of clot lysed after 30 minutes, and (E) time until complete clot lysis; n = 3 mice per treatment group. (F-G) Western blot against FXIII-A and TEG curve tracing of plasma from untreated (Unt) mice (lavender), mice pretreated with siFXIIIB (green), and mice pretreated with siFXIIIB and given an IV dose of mFXIII (yellow). (H-I) Representative western blots against (H) fibrin(ogen) and (I) α2-antiplasmin comparing the HMW species in plasma clotted for 1 hour from untreated mice or those treated with siFXIIIB 2 weeks previously. Clots were formed and lysed in plasma samples with platelets (PRP) or without platelets (PPP) 1 hour after adding CaCl2 to plasma. Non-recalcified plasma was used as a control for unactivated (UA) FXIII, and plasma with an inhibitor of FXIII-A* was added in vitro as a control (T101, 0.8 mM). (J-K) Crosslinking of fibrin and α2-antiplasmin was assessed over time by western blot of PPP clots from untreated mice (lavender) or siFXIIIB pretreated mice (green) and quantified using densitometry. Graphs show the percentage of fibrin or α2-antiplasmin found in the HMW range (100-200 kDa) compared with the total amount of fibrin or α2-antiplasmin. For all graphs, values represent mean ± SEM. **P < .01, and ns indicates not significant.
siFXIIIB delays crosslinking of α2-antiplasmin, leading to clots that are more susceptible to lysis ex vivo. (A-B) Representative TEG curve tracings using blood from mice treated with siLuc (A, lavender) or with siFXIIIB (B, green). In each case, recombinant tissue factor was added to stimulate clotting and 4 nM of tPA was added to enhance lysis. (C-E) TEG curves were quantified for (C) maximum clot stiffness, (D) percent of clot lysed after 30 minutes, and (E) time until complete clot lysis; n = 3 mice per treatment group. (F-G) Western blot against FXIII-A and TEG curve tracing of plasma from untreated (Unt) mice (lavender), mice pretreated with siFXIIIB (green), and mice pretreated with siFXIIIB and given an IV dose of mFXIII (yellow). (H-I) Representative western blots against (H) fibrin(ogen) and (I) α2-antiplasmin comparing the HMW species in plasma clotted for 1 hour from untreated mice or those treated with siFXIIIB 2 weeks previously. Clots were formed and lysed in plasma samples with platelets (PRP) or without platelets (PPP) 1 hour after adding CaCl2 to plasma. Non-recalcified plasma was used as a control for unactivated (UA) FXIII, and plasma with an inhibitor of FXIII-A* was added in vitro as a control (T101, 0.8 mM). (J-K) Crosslinking of fibrin and α2-antiplasmin was assessed over time by western blot of PPP clots from untreated mice (lavender) or siFXIIIB pretreated mice (green) and quantified using densitometry. Graphs show the percentage of fibrin or α2-antiplasmin found in the HMW range (100-200 kDa) compared with the total amount of fibrin or α2-antiplasmin. For all graphs, values represent mean ± SEM. **P < .01, and ns indicates not significant.
FXIII-A* inhibits fibrinolysis by forming intra- and intermolecular crosslinks between fibrin monomers and antifibrinolytic proteins such as α2-antiplasmin during coagulation.49,50 These form high molecular weight (HMW) adducts that are stable in detergent. To assess how siFXIIIB knockdown impacts the crosslinking of clots, fibrin and α2-antiplasmin were analyzed by western blot in plasma collected from mice and clotted in vitro (Figure 2H-I). Recalcified platelet poor plasma (PPP) and PRP had HMW fibrin and α2-antiplasmin products in samples from untreated mice after 1 hour of clotting. Unactivated plasma and plasma containing FXIII inhibitor T101 had only small amounts of these HMW products. Levels of HMW fibrin and α2-antiplasmin products >100 kDa were lower in PPP collected from mice treated with siFXIIIB compared with untreated mice. In the siFXIIIB samples, PRP had more HMW fibrin than the PPP equivalent. In untreated PPP, maximal crosslinking was completed within 1 hour, but crosslinking of fibrin and α2-antiplasmin continued in PPP from mice treated with siFXIIIB for 1 to 24 hours (Figure 2J-K). Combined, these experiments show that knockdown of plasma FXIII impaired crosslinking of α2-antiplasmin to fibrin and enhanced fibrinolysis without substantially inhibiting clot formation.
A specific siRNA sequence was also designed to knock down FXIII-B mRNA in rabbits (siFXIIIB-r). FXIII-A was depleted by 90% in plasma 11 days after a single administration and began returning to baseline in the sixth week (Figure 3A-B). When rabbit plasma was clotted ex vivo, there was a significant decrease of HMW α2-antiplasmin in siFXIIIB-r plasma compared with siLuc plasma (Figure 3C). HMW fibrin was also decreased (supplemental Figure 3). There were no HMW products in the 2 negative controls, which were unactivated plasma without additional Ca2+ and activated plasma containing T101.

Knockdown of FXIII-B in rabbits. (A) Western blots against FXIII-A. Each lane contains the plasma from an individual rabbit treated with siFXIIIB-r (green) or siLuc (lavender). (B) Quantification of FXIII-A protein from panel A using densitometry. Values represent mean ± range; n = 2. (C) Western blot against α2-antiplasmin comparing HMW species in plasma clotted for 1 hour that had been collected 14 or 21 days after rabbits were treated with siFXIIIB-r or siLuc. Non-recalcified plasma was used as a control for unactivated FXIII-A.
Knockdown of FXIII-B in rabbits. (A) Western blots against FXIII-A. Each lane contains the plasma from an individual rabbit treated with siFXIIIB-r (green) or siLuc (lavender). (B) Quantification of FXIII-A protein from panel A using densitometry. Values represent mean ± range; n = 2. (C) Western blot against α2-antiplasmin comparing HMW species in plasma clotted for 1 hour that had been collected 14 or 21 days after rabbits were treated with siFXIIIB-r or siLuc. Non-recalcified plasma was used as a control for unactivated FXIII-A.
Fibrinolysis was enhanced by siFXIIIB in an in vivo model of thrombolysis
To investigate thrombus stability in vivo, a model of thrombosis and reperfusion with a tPA analog (TNK) was used.45 To induce thrombosis, FeCl3 was applied to exposed carotid arteries of C57BL/6J mice, while blood flow was measured by Doppler ultrasound. Once the carotid artery was occluded by the thrombus, TNK was administered intravenously at 9 mg per kg of body weight, which is approximately half the therapeutic dose for humans. Blood flow recovered to a greater extent in mice treated with siFXIIIB (P = .0015), compared with mice that received siLuc (Figure 4A-C). At this relatively low dose of TNK, 8 of 9 mice treated with siLuc had no reperfusion, and 1 had unstable intermittent reperfusion. In contrast, 2 of 9 mice treated with siFXIIIB had complete reperfusion, 4 of the 9 mice had unstable intermittent perfusion and reperfusion, and 3 had no reperfusion.
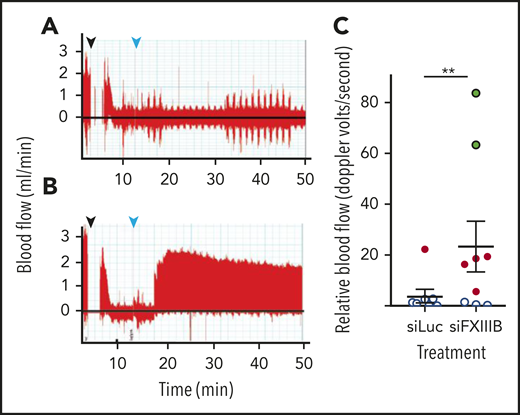
Pretreatment with siFXIIIB renders arterial thrombi more susceptible to lysis in vivo. Doppler ultrasound measured blood flow after thrombi were induced in the carotid artery with a 10% w/v solution of FeCl3, and tenecteplase was administered at a dose of 9 mg/kg. (A-B) Representative Doppler graphs show the time when FeCl3 was applied (black arrow, followed by a 2-minute gap in measurement), occlusion of the vessel, drop in blood flow, injection of tenecteplase (light blue arrow), and then either no recovery of blood flow (A, siLuc treated) or stable reperfusion (B, siFXIIIB treated). (C) Quantification of panels A and B, measuring the blood flow until 60 minutes after occlusion (n = 9). Data markers indicate whether reperfusion was stable (green), transient (red), or if no reperfusion occurred (white). For all graphs, values represent mean ± SEM. ns, P > .05; **P < .01.
Pretreatment with siFXIIIB renders arterial thrombi more susceptible to lysis in vivo. Doppler ultrasound measured blood flow after thrombi were induced in the carotid artery with a 10% w/v solution of FeCl3, and tenecteplase was administered at a dose of 9 mg/kg. (A-B) Representative Doppler graphs show the time when FeCl3 was applied (black arrow, followed by a 2-minute gap in measurement), occlusion of the vessel, drop in blood flow, injection of tenecteplase (light blue arrow), and then either no recovery of blood flow (A, siLuc treated) or stable reperfusion (B, siFXIIIB treated). (C) Quantification of panels A and B, measuring the blood flow until 60 minutes after occlusion (n = 9). Data markers indicate whether reperfusion was stable (green), transient (red), or if no reperfusion occurred (white). For all graphs, values represent mean ± SEM. ns, P > .05; **P < .01.
Knockdown of FXIII-B increased re-bleeds but not blood loss in an in vivo bleeding model
A model of bleeding by tail transection was used to assess hemostasis in mice treated with siFXIIIB (Figure 5A). After tail transection, re-bleeds were counted during the 40-minute observation period, and total blood lost was collected and quantified by measuring hemoglobin spectrophotometrically. Endogenous hemostasis in mice treated with siFXIIIB halted the bleeding, but wounds were more prone to spontaneous re-bleeding (Figure 5B). This resulted in a significantly greater number of bleeding events as defined by a preceding cessation of bleeding for longer than 20 seconds. Overall, mice treated with siFXIIIB had amounts of total blood loss similar to those of untreated mice during the 40-minute observation period (Figure 5C). Mice given a therapeutic dose of the FXa inhibitor apixaban before tail transection had significantly impaired clotting compared with mice treated with siFXIIIB (P = .005) and untreated mice (P = .007). No spontaneous or excessive bleeding was apparent in siFXIIIB-treated mice during the 5 months of sustained FXIII-B knockdown. These findings show that although knockdown of FXIII-B mediated a decrease in plasma FXIII-A and destabilized clots, it did not lead to excess blood loss.
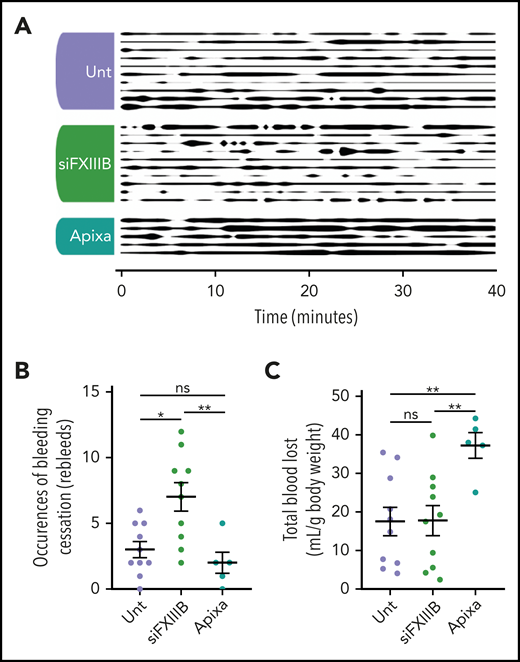
Knockdown of FXIII-B results in more frequent re-bleeds but not an increase in blood loss in an in vivo model of bleeding. Tails of mice pretreated with apixaban (Apixa), siFXIIIB (siFXIII), or untreated (Unt) were transected and the subsequent bleed was observed over a period of 40 minutes. (A) Graphical representation of bleeding from the wound. Line thickness corresponds to bleeding severity, observed at 60-second intervals and qualitatively rated on a scale of 0 to 5: 0 (no line) is a cessation of bleeding, and 5 (thick line) is full unmitigated bleeding. (B) The number of clotting events, defined as a cessation of bleeding for 20 seconds or longer. (C) The total volume of blood lost relative to the individual mouse’s weight was quantified by spectrophotometry measuring hemoglobin. Each row (A) or point (B- C) represents 1 animal. Data are presented as the mean ± SEM. ns, P > .05; *P < .05; **P < .01.
Knockdown of FXIII-B results in more frequent re-bleeds but not an increase in blood loss in an in vivo model of bleeding. Tails of mice pretreated with apixaban (Apixa), siFXIIIB (siFXIII), or untreated (Unt) were transected and the subsequent bleed was observed over a period of 40 minutes. (A) Graphical representation of bleeding from the wound. Line thickness corresponds to bleeding severity, observed at 60-second intervals and qualitatively rated on a scale of 0 to 5: 0 (no line) is a cessation of bleeding, and 5 (thick line) is full unmitigated bleeding. (B) The number of clotting events, defined as a cessation of bleeding for 20 seconds or longer. (C) The total volume of blood lost relative to the individual mouse’s weight was quantified by spectrophotometry measuring hemoglobin. Each row (A) or point (B- C) represents 1 animal. Data are presented as the mean ± SEM. ns, P > .05; *P < .05; **P < .01.
Discussion
A specific inhibitor of FXIII-A* that is suitable for use in multiple species of experimental animals as well as humans has not yet been developed, despite its potential therapeutic and experimental value. To address this need, we used an siRNA-LNP strategy that inhibited hepatic FXIII-B mRNA expression and resulted in sustained and specific depletion of plasma FXIII-B and FXIII-A2. Plasma-derived FXIII-A2 was depleted by more than 90% in mice and rabbits, and the depletion lasted for weeks after a single administration of siFXIIIB. The decrease in FXIII-A was similar to or greater than the decrease in FXIII-B. FXIII-B is normally present in a twofold excess over FXIII-A, and patients with FXIII-B deficiency can still have more FXIII-B than FXIII-A.32 These data are consistent with an equilibrium of free and complexed FXIII-B, with the free B subunit present in both monomeric and dimeric forms.6,51
With repeated dosing of mice at 3-week intervals, FXIII-A knockdown was sustained for more than 5 months without any observed adverse effects. Previous pharmacologic approaches have been limited by agents that lack specificity52 or exhibit short half-lives; thus only in vivo studies of FXIII-A inhibition with constant infusion of the inhibitor are possible.21,53 Repeated dosing with siRNA-LNPs is used in humans for long-term treatment of hereditary transthyretin-mediated amyloidosis.42 IV administration of exogenous FXIII rapidly reversed the effects of siFXIIIB-mediated knockdown by restoring FXIII-A* activity, suggesting that knockdown could also be readily reversed by plasma transfusion. This reversibility is consistent with previous reports with FXIII-deficient mice54 and is an approach used in patients with congenital FXIII deficiency.55 Cellular FXIII-A2 was protected from the siFXIIIB-mediated decrease of FXIII-A because it does not depend on the stabilizing effect of the tetrameric arrangement of FXIII-A2B2. This indicates that siFXIIIB can be used to distinguish the contribution of plasma-derived FXIII-A2 from cellular FXIII-A2 in biological and pathological experimental models. This tool can enable knockdown of FXIII in most disease models, except for those that cause impaired hepatocyte uptake of LNPs, such as Ldlr knockouts. Thus, siFXIIIB may be useful to further investigate the role of FXIII-A2B2 in animal models of disease, with potential translation to humans.
Additional approaches to prevent and alleviate thrombosis are needed, as highlighted by the challenges in using current antithrombotics for COVID-19–associated coagulopathy.15,16 An approach that specifically enhances endogenous fibrinolysis may be useful for decreasing the incidence and burden of thrombosis without incurring significant bleeding risk. Even though inhibitors of thrombin activatable finbrinolysis inhibitor (TAFI), an enzyme that acts as an antifibrinolytic among other functions, have been evaluated in clinical trials, approaches to specifically modulate fibrinolysis through inhibition of antifibrinolysis are limited. The results of our study show that siRNA-mediated depletion of FXIII-B and subsequent diminution of plasma FXIII-A led to clots with increased susceptibility to lysis ex vivo and in vivo because of impaired crosslinking of α2-antiplasmin to fibrin. Fibrin crosslinking was not abolished completely and was only moderately impaired in the presence of platelets, and so clot formation and hemostasis were not significantly compromised. The increased amount of HMW fibrin in PRP samples was likely the result of the fibrin binding and facilitating surface provided by the platelets.14,56 Although mice treated with siFXIIIB were more prone to re-bleeding after tail transection, total blood loss was not significantly increased because clots reformed quickly. This is consistent with a previous finding that blocking the interaction between plasma FXIII-A2B2 and fibrinogen does not lead to a hemostatic defect in mice.13 This differs for FXIII-A knockout mice, which bleed more than wild-type mice.57 This critical difference is likely a result of residual plasma FXIII-A2 and platelet-derived FXIII-A2 in siFXIIIB-treated mice. These results are consistent with the phenotypes of patients who are deficient in FXIII-B who typically have a milder bleeding diathesis than patients with FXIII-A deficiency.30 If siFXIIIB is to be developed into a therapeutic that enhances thrombolysis and decreases thrombosis through enhanced fibrinolysis, more work will be needed to assess the safety of this approach. In a mouse model of deep vein thrombosis induced by FeCl3, FXIII-A knockout mice had increased pulmonary embolism, similar to dabigatran-enhanced pulmonary embolism.58 The amount of FXIII-A needed to prevent embolization has not been established, but pulmonary embolism is not a reported feature in people deficient in FXIII-B.30
In summary, using single bolus injection of siRNA to knock down the expression of the FXIII-B subunit is highly effective at reducing the plasma reservoir of FXIII-A2, while leaving cellular FXIII-A2 unaffected. Targeting the plasma fraction of FXIII-A2 for prophylactic attenuation is advantageous to sufficiently delay the procoagulant response for prolonged periods without overt bleeding. Further work is needed to establish the utility of FXIII-B knockdown in controlling fibrinolysis and decreasing thrombosis in humans. The preclinical work presented here has helped to establish a framework for investigating the functions of FXIII-A2B2 in combination with diverse implicated pathologies.
For original data, please contact ckastrup@msl.ubc.ca.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by grants from the Canadian Institutes of Health Research (CIHR) (FDN-148370, MSH-130166), the Natural Sciences and Engineering Research Council (NSERC) (RGPIN 2018-04918), the Michael Smith Foundation for Health Research (16498), the Canadian Foundation for Innovation (31928) (C.J.K., A.W.S., J.L.), and the Canadian Venous Thromboembolism Clinical Trials and Outcomes Research (CanVECTOR) Network (A.W.S.). E.L.G.P., S.C.M., and M.R.S. were supported by a grant from the Heart and Stroke Foundation of Canada (HSFC) (G-19-0026524). E.M.C. was supported by the Canada Research Chairs and the CIHR, NSERC, and HSFC.
Authorship
Contribution: A.W.S. designed and performed most experiments, analyzed and interpreted the data, made the figures, and wrote the article; S.C.M., J.L., and N.S.S. helped perform experiments and analyzed and interpreted the data; J.A.K., H.M.R., R.v.d.M., and M.R.S. helped design and execute the experiments; A.P.O., J.S.P., E.M.C., E.L.G.P., and P.R.C., helped design experiments, analyze data, and edit the article; and C.J.K. designed experiments, interpreted the data, and wrote the article.
Conflict-of-interest disclosure: P.R.C. acknowledges financial interests in Acuitas Therapeutics and Precision NanoSystems. The remaining authors declare no competing financial interests.
Correspondence: Christian J. Kastrup, Michael Smith Laboratories and Department of Biochemistry and Molecular Biology, University of British Columbia, 2185 East Mall, Vancouver, BC, Canada V6T 1Z4; e-mail: ckastrup@msl.ubc.ca.
