

TO THE EDITOR:
Chronic myelomonocytic leukemia (CMML), an aggressive hematological neoplasm characterized by sustained peripheral blood monocytosis (≥1 × 109/L, ≥10% of white cell count), is associated with a median overall survival of 28 to 32 months, with no current therapies that improve its natural history.1,2 Although hypomethylating agents (HMAs) have been approved for the management of CMML, the overall response and complete response rates are 40% to 50% and <20% respectively, with no impact on mutational allele burdens, even in responding patients.3,4 We have previously demonstrated that primary CMML samples exhibit granulocyte-macrophage colony-stimulating factor (GM-CSF)–dependent hypersensitivity by hematopoietic progenitor colony-formation assays and by phospho-STAT5 (pSTAT5) flow cytometry and that the GM-CSF axis is a viable therapeutic target in CMML.5 Lenzilumab (KB003) is a novel engineered human immunoglobulin G1κ monoclonal antibody, with high affinity for human GM-CSF, that has activity in preclinical models of CMML.5 In addition, from a safety perspective, lenzilumab and its precursor, KB002 (chimeric version), have been tested in multiple single- and repeat-dose phase 1-2 clinical trials evaluating IV administration of 0.2 to 10 mg/kg (or flat dosing of 400 and 600 mg) with no deaths, drug-related serious adverse events (AEs), or withdrawals due to AEs. The largest published study to date randomized 160 patients with inadequately controlled bronchial asthma to receive lenzilumab (n = 78) or placebo (n = 82), with no grade 3-4 drug-related AEs.6 Notably, there were no changes in biomarkers (serum surfactant protein D) or clinical/radiological features of pulmonary alveolar proteinosis, a known complication with anti–GM-CSF monoclonal antibody therapy.6 We report a phase 1 clinical trial testing the safety and preliminary efficacy of single-agent lenzilumab in CMML patients who were refractory, intolerant, or deemed ineligible for HMA or hydroxyurea therapy (only 20% of patients in this trial were treatment naive).
The study was approved by scientific and ethical review boards at the Mayo Clinic and at the Moffitt Cancer Center. All patients provided written informed consent to participate in the study. This was a multicenter phase 1 study designed to evaluate the safety and determine the recommended phase 2 dose of lenzilumab in subjects with CMML (NCT02546284). Dose escalation proceeded using a standard 3+3 study design to determine the maximum tolerated dose, with 6 evaluable patients required at the maximum tolerated dose (supplemental Figure 1, available on the Blood Web site). The 3 dose cohorts included 200 mg, 400 mg, and 600 mg given IV on days 1 and 15 of cycle 1 and then on day 1 of subsequent 28-day cycles. Key inclusion criteria included a World Health Organization (WHO)-defined diagnosis of CMML, an absolute neutrophil count >0.5 × 109/L, and a platelet count >20 × 109/L (supplemental Table 1, study inclusion/exclusion criteria).7 Response was evaluated utilizing the 2015 myelodysplastic syndrome (MDS)/myeloproliferative neoplasm (MPN) International Working Group (IWG)’s response criteria.8 Pharmacokinetics analysis and pharmacodynamics were evaluated by pSTAT5 using flow cytometry at screening and at day 1, cycle 3. Next-generation sequencing was carried out for myeloid-relevant genes on bone marrow mononuclear cells at screening and at day 1, cycle 3. Progenitor colony-forming assays were carried out in select patients, with the same cells also being used to generate patient-derived xenografts (PDXs; NSG-SGM3 mice), by previously described methods.9 Because of financial limitations, we were not able to assess for anti–GM-CSF antibody generation in study subjects, although serial samples have been collected, and there is a plan to complete this at a later date.
Between July of 2016 and June of 2018, 15 patients with WHO-defined CMML were enrolled. The median age at study entry was 74 years (range, 52-85 years), and 80% were male (Table 1; supplemental Table 2). Nine (60%) patients were classified as CMML-0, and 3 (20%) patients each were classified as CMML-1 or CMML-2. Seventy-three percent of patients had normal cytogenetics or loss of chromosome Y. The most commonly mutated genes at screening included TET2 (60%), ASXL1 (53%), and SRSF2 (47%); RAS pathway (ie, NRAS or CBL) mutations were found in 40% of patients (Figure 1A; Table 1). Nine (60%) patients were previously treated with HMAs and/or experimental therapies, 3 (20%) were treated with hydroxyurea, and 3 (20%) were untreated, because they were deemed to be unlikely to respond to HMAs or hydroxyurea. The median hemoglobin at enrollment was 9.7 g/dL (range, 7.6-14 g/dL), median white blood cell count was 10 × 109/L (range, 5.3-59.8 × 109/L), median absolute monocyte count was 4.5 × 109/L (range, 1.3-10 × 109/L), median platelet count was 147 × 109/L (range, 16-942 × 109/L), and 66% of cases were subclassified as MPN-CMML by the French-American-British classification at study entry.7 None of the study patients had eosinophilia or basophilia prior to enrollment in the study. Three patients were enrolled at each dose level, and 6 additional patients were enrolled at the 600-mg dose, as planned. No dose-limiting toxicities were identified, and no treatment-emergent study drug–related grade 3 or 4 toxicities were reported (Figure 1B). In total, 1 patient each had grade 2 anemia and neutropenia, whereas 2 and 1 patients had grade 3 and grade 4 neutropenia (not attributable to the study drug), respectively. Pulmonary alveolar proteinosis has been associated with the development of anti–GM-CSF antibodies; however, no case of pulmonary alveolar proteinosis was seen in this study cohort. The mean duration on therapy was 110 days (range, 14-798), and the majority of patients discontinued study drug because of disease progression or lack of response (69%). Median duration of best response was 5.5 months (range, 1-14 months) (Figure 1A). Four of 15 (27%) patients enrolled achieved clinical benefit by MDS/MPN IWG criteria, with 3 platelet responses and 1 neutrophil response. An additional patient had bone marrow myeloblast reduction from 6% to 1% (partial marrow response), which allowed him to undergo allogeneic hematopoietic stem cell transplantation. The overall cumulative response rate (excluding stable disease) was 33.33%. Although 1 patient did have a reduction in splenomegaly, it did not meet IWG criteria for a spleen response, and he was classified as having stable disease. Three of 5 (60%) responding patients had NRAS mutations, whereas only 1 of 10 (10%) nonresponding patients had an NRAS mutation (Figure 1A). This is consistent with prior reports suggesting that GM-CSF hypersensitivity in CMML is more prominent in patients with RAS pathway mutations.5 Clinical responses did not correlate with changes in pSTAT5 between screening and cycle 3 (supplemental Figure 2), and there were no changes in mutational allele burdens in responding patients (supplemental Figure 3). Although GM-CSF signals through JAK2 and STAT5 in CMML, STAT5 isoform expression, STAT5 activation, and STAT5 target gene expression are altered significantly; further studies are needed to understand the correct timing of these assessments.10 Hematopoietic progenitor colony-forming assays were carried out in 3 patients, including 1 responder (NRAS mutant) and 2 nonresponders (NRAS wild-type); marked GM-CSF hypersensitivity was observed in the responding patient vs the nonresponders (supplemental Figure 4). We were also able to generate PDXs from 2 patient samples (NRAS mutant responder vs NRAS wild-type nonresponder) and show that the responding patient sample resulted in more robust bone marrow and spleen engraftment (supplemental Figure 5a), as well as more prominent splenomegaly (supplemental Figure 5b), in comparison with the nonresponder, providing us with potential biomarkers for accurate patient selection. Of note, GM-CSF hypersensitivity and robust PDX engraftment are innate characteristics of RAS pathway–mutant CMML.
Clinical and genetic information about CMML patients enrolled in the KB003 clinical trial
| Patient | Age, y | Sex | CMML Classification | Cytogenetics | Molecular abnormalities | Prior therapies | Maximum KB003 dose, mg* | Cycles given, n | Best response | Duration of best response, mo | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| WHO | FAB | ||||||||||
| 1 | 73 | M | CMML-1 | Proliferative | 45,X,-Y | ASXL1, CBL, IDH2, SRSF2 | Hydroxyurea | 200 | 4 | None | — |
| 2 | 77 | M | CMML-0 | Proliferative | 46,XY,t(3;12;6)(p21;q21;q23)[8]/46,XY[12] | ASXL1, EZH2, TET2 | Hydroxyurea | 200 | 2 | None | — |
| 3 | 52 | M | CMML-0 | Dysplastic | 46,XY | SRSF2, TET2 | None | 600 (initially enrolled in 200-mg cohort) | 25 | Clinical benefit (platelet response) | 24 |
| 4 | 80 | M | CMML-0 | Proliferative | 46,XY | SRSF2, TET2 | Darbepoietin alfa, investigational agent (clinical trial), decitabine, investigational agent (clinical trial) | 400 | 6 | Stable disease | 6 |
| 5 | 74 | F | CMML-0 | Proliferative | 46,XX,del(20)(q11.2q13.1) [17]/46,XX [3] | ASXL1, NRAS, TET2 | Azacitidine, lenalidomide, hydroxyurea, decitabine, investigational agent (clinical trial) | 600 (initially enrolled in 400-mg cohort) | 29 | Stable disease | 26 |
| 6 | 68 | M | CMML-0 | Dysplastic | 46,XY | ASXL1, BCOR, EZH2, SF3B1 | Azacitidine | 600 | 2 | None | — |
| 7 | 66 | M | CMML-0 | Proliferative | 46,XY | ASXL1, NRAS, SRSF2, TET2 | Investigational agent (clinical trial) | 600 | 9 | Clinical benefit (platelet response) | 7 |
| 8 | 71 | M | CMML-0 | Proliferative | 45,X,-Y | ASXL1, NRAS, RUNX1 | None | 600 | 11 | Clinical benefit (platelet response) | 10 |
| 9 | 79 | M | CMML-2 | Proliferative | 46,XY | Not available | Hydroxyurea | 600 | 1 | None | — |
| 10 | 62 | M | CMML-2 | Dysplastic | 46,XY | CBL, TET2 | Decitabine | 600 | 1 | None | — |
| 11 | 68 | M | CMML-1 | Proliferative | 46,XY | ASXL1, NRAS, SRSF2, TET2 | None | 400 | 4 | Partial marrow response | 3 |
| 12 | 85 | M | CMML-0 | Dysplastic | 46,XY | TET2, SRSF2 | Decitabine | 600 | 4 | Stable disease | 3 |
| 13 | 76 | M | CMML-2 | Proliferative | 45,-7,XY | ASXL1, SRSF2 | Azacitidine, decitabine | 600 | 2 | Stable disease | 1 |
| 14 | 77 | M | CMML-1 | Dysplastic | 46, XX | TET2, EZH2, SETBP1, RUNX1 | Azacitidine | 600 | 4 | Clinical benefit (neutrophil response) | 6 |
| 15 | 71 | F | CMML-0 | Proliferative | 46, XX | TET2, SF3B1, JAK2 | Ruxolitinib, decitabine, azacitidine | 600 | 14 | Clinical benefit (spleen response) | 14 |
| Patient | Age, y | Sex | CMML Classification | Cytogenetics | Molecular abnormalities | Prior therapies | Maximum KB003 dose, mg* | Cycles given, n | Best response | Duration of best response, mo | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| WHO | FAB | ||||||||||
| 1 | 73 | M | CMML-1 | Proliferative | 45,X,-Y | ASXL1, CBL, IDH2, SRSF2 | Hydroxyurea | 200 | 4 | None | — |
| 2 | 77 | M | CMML-0 | Proliferative | 46,XY,t(3;12;6)(p21;q21;q23)[8]/46,XY[12] | ASXL1, EZH2, TET2 | Hydroxyurea | 200 | 2 | None | — |
| 3 | 52 | M | CMML-0 | Dysplastic | 46,XY | SRSF2, TET2 | None | 600 (initially enrolled in 200-mg cohort) | 25 | Clinical benefit (platelet response) | 24 |
| 4 | 80 | M | CMML-0 | Proliferative | 46,XY | SRSF2, TET2 | Darbepoietin alfa, investigational agent (clinical trial), decitabine, investigational agent (clinical trial) | 400 | 6 | Stable disease | 6 |
| 5 | 74 | F | CMML-0 | Proliferative | 46,XX,del(20)(q11.2q13.1) [17]/46,XX [3] | ASXL1, NRAS, TET2 | Azacitidine, lenalidomide, hydroxyurea, decitabine, investigational agent (clinical trial) | 600 (initially enrolled in 400-mg cohort) | 29 | Stable disease | 26 |
| 6 | 68 | M | CMML-0 | Dysplastic | 46,XY | ASXL1, BCOR, EZH2, SF3B1 | Azacitidine | 600 | 2 | None | — |
| 7 | 66 | M | CMML-0 | Proliferative | 46,XY | ASXL1, NRAS, SRSF2, TET2 | Investigational agent (clinical trial) | 600 | 9 | Clinical benefit (platelet response) | 7 |
| 8 | 71 | M | CMML-0 | Proliferative | 45,X,-Y | ASXL1, NRAS, RUNX1 | None | 600 | 11 | Clinical benefit (platelet response) | 10 |
| 9 | 79 | M | CMML-2 | Proliferative | 46,XY | Not available | Hydroxyurea | 600 | 1 | None | — |
| 10 | 62 | M | CMML-2 | Dysplastic | 46,XY | CBL, TET2 | Decitabine | 600 | 1 | None | — |
| 11 | 68 | M | CMML-1 | Proliferative | 46,XY | ASXL1, NRAS, SRSF2, TET2 | None | 400 | 4 | Partial marrow response | 3 |
| 12 | 85 | M | CMML-0 | Dysplastic | 46,XY | TET2, SRSF2 | Decitabine | 600 | 4 | Stable disease | 3 |
| 13 | 76 | M | CMML-2 | Proliferative | 45,-7,XY | ASXL1, SRSF2 | Azacitidine, decitabine | 600 | 2 | Stable disease | 1 |
| 14 | 77 | M | CMML-1 | Dysplastic | 46, XX | TET2, EZH2, SETBP1, RUNX1 | Azacitidine | 600 | 4 | Clinical benefit (neutrophil response) | 6 |
| 15 | 71 | F | CMML-0 | Proliferative | 46, XX | TET2, SF3B1, JAK2 | Ruxolitinib, decitabine, azacitidine | 600 | 14 | Clinical benefit (spleen response) | 14 |
F, female; FAB, French-American-British classification; M, male.
Given every 28 days.
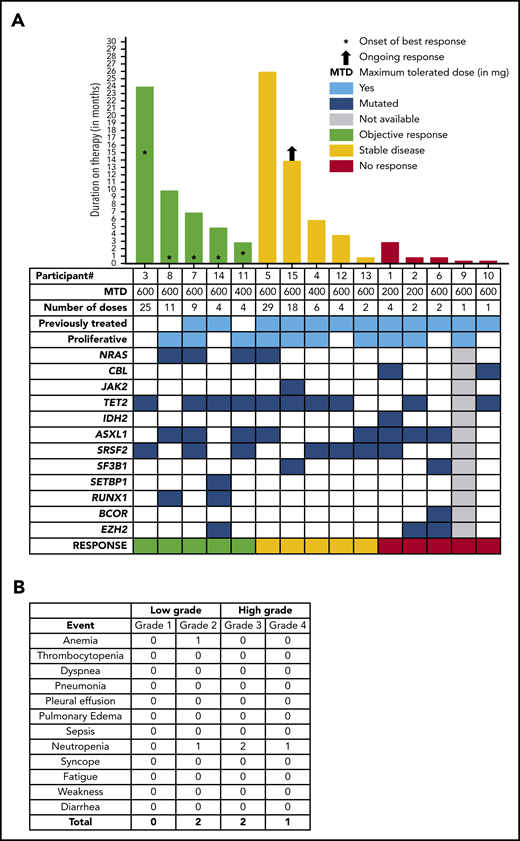
Clinical and response characteristics of 15 patients with CMML enrolled in the phase 1 lenzilumab study. (A) Oncoplot demonstrating the genotype of 15 CMML patients as documented at the time of enrollment into the study and their correlation with disease responses, as adjudicated by the IWG MDS/MPN overlap syndrome response criteria. It also shows the dose cohort, number of doses of the study drug, exposure to prior therapies, and duration of response. The patient number (as indicated in Table 1) is shown below the x-axis. (B) Grades 1-4 AEs encountered in the phase 1 lenzilumab CMML study.
Clinical and response characteristics of 15 patients with CMML enrolled in the phase 1 lenzilumab study. (A) Oncoplot demonstrating the genotype of 15 CMML patients as documented at the time of enrollment into the study and their correlation with disease responses, as adjudicated by the IWG MDS/MPN overlap syndrome response criteria. It also shows the dose cohort, number of doses of the study drug, exposure to prior therapies, and duration of response. The patient number (as indicated in Table 1) is shown below the x-axis. (B) Grades 1-4 AEs encountered in the phase 1 lenzilumab CMML study.
In summary, we report the first study of lenzilumab in CMML and document that the drug is well tolerated, without any drug-related grade 3-4 AEs. Durable clinical benefit was achieved in 33% of patients, and 1 patient with a partial marrow response was bridged to an allogeneic hematopoietic stem cell transplantation, providing proof of concept that GM-CSF inhibition is a viable therapeutic strategy in CMML. Responses were seemingly better in RAS pathway–mutant CMML patients and in those who demonstrated GM-CSF hypersensitivity in colony-forming assays and robust PDX engraftment (both are innate features of RAS pathway–mutant CMML). Similar GM-CSF hypersensitivity is seen in juvenile myelomonocytic leukemia, a pediatric MDS/MPN overlap neoplasm that is driven largely by germline and somatic RAS pathway mutations (bona fide RASopathy), with poor outcomes.11 Future studies are warranted to identify CMML patient subtypes who are more likely to respond (eg, CMML patients with RAS pathway mutations and/or those with GM-CSF hypersensitivity in colony-forming assays), to identify rational combination strategies and to test the safety and efficacy of lenzilumab in children with juvenile myelomonocytic leukemia.
Contact the corresponding author for original data.
The online version of this article contains a data supplement.
Acknowledgment
The authors thank all of the patients who took part in the study.
Authorship
Contribution: M.M.P., D.A.S., A.A.M., and E.P. designed the study and enrolled subjects; R.H., J.H., D.Z., A.A.-K., M.B., N.G., R.S.K., A.T., and A.F.L. enrolled subjects; H.N., T.L.K., C.L., and M.E.B. performed correlative analyses; and A.L., T.S., and C.D. represent the pharmaceutical company that helped with trial design and sponsorship.
Conflict-of-interest disclosure: M.M.P. has served on the advisory board for Stemline Therapeutics. A.F.L. has received research funding from and has served as a consultant for Celgene and serves as a scientific advisor for Cellular Biomedicine Group and Aileron Therapeutics, Inc. E.P. has received research funding from Humanigen Inc. (formerly KalaBios Pharmaceuticals), Incyte, Celgene, and Kura and has served on advisory boards for Stemline Therapeutics and Blueprint Medicines. The remaining authors declare no competing financial interests.
Correspondence: Eric Padron, Malignant Hematology, Moffitt Cancer Center, 12902 Magnolia Dr, Tampa, FL 33612; e-mail: eric.padron@moffitt.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal