

In this issue of Blood, have reliably defined a previously elusive entity, that of blastic plasmacytoid dendritic cell neoplasm (BPDCN)-like acute myeloid leukemia (AML), or pDC-AML, which occurs in ∼5% of AML cases. Remarkably, in the vast majority of cases, pDC-AML is associated with occurrence of somatic mutations of RUNX1.1
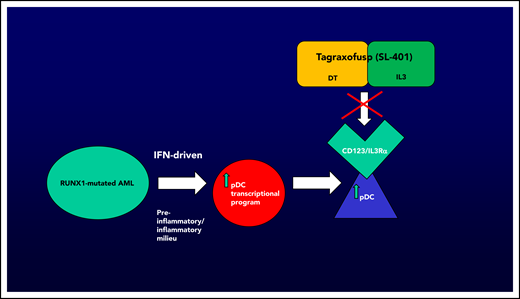
AML with pDC expansion, or pDC-AML, is associated with RUNX1 mutations in the vast majority of cases (∼70%). Xiao et al demonstrate that blasts from pDC-AML that harbor RUNX1 mutations may acquire an interferon-driven pDC transcriptional program, thereby leading to increased pDCs. pDCs are known to express/overexpress CD123, or IL3Rα, which can uniquely be targeted by CD123-directed therapy. Tagraxofusp (SL-401, DT-IL3) is the first approved agent targeting CD123 (BPDCN, age ≥2 years, December 2018), opening up the possibility of novel avenues for therapeutic investigation. Figure by N. Pemmaraju.
AML with pDC expansion, or pDC-AML, is associated with RUNX1 mutations in the vast majority of cases (∼70%). Xiao et al demonstrate that blasts from pDC-AML that harbor RUNX1 mutations may acquire an interferon-driven pDC transcriptional program, thereby leading to increased pDCs. pDCs are known to express/overexpress CD123, or IL3Rα, which can uniquely be targeted by CD123-directed therapy. Tagraxofusp (SL-401, DT-IL3) is the first approved agent targeting CD123 (BPDCN, age ≥2 years, December 2018), opening up the possibility of novel avenues for therapeutic investigation. Figure by N. Pemmaraju.
Clearly defining pDC-AML has been extremely difficult. Xiao et al found that pDC-AML carries its own immune signature; one that is intermediate between BPDCN and AML. In comparison with BPDCN, pDC-AML also has a 3:1 male predominance, greater tendency for skin involvement, and poor clinical outcomes; in contrast to BPDCN, it is CD34+, not as commonly CD56+, and very commonly !RUNX1 mutated (70+% vs <5% in BPDCN), thereby convincingly making this a distinct entity.
The elucidation of the connection of RUNX1 mutations and a malignant pDC transcriptional program by Xiao et al is quite significant. The confirmation of the importance of RUNX1, coupled with the known importance of CD123 and TCF4’s critical role,2,3 adds to the developing mechanistic story of regulation and propagation of the pDC malignant state. The demonstration that RUNX1 is a key regulator of an inflammatory/interferon-response pDC transcriptional program that leads to increased numbers of pDCs, known to express CD123 (or IL3Rα), suggests that CD123-targeted agents are worth investigating (see figure). Xiao et al tested this hypothesis in xenograft models of pDC-AML, demonstrating that targeting with tagraxofusp (first-in-class CD123 targeting agent) led to elimination of malignant pDCs and a two- to threefold decrease in leukemia tumor burden, but not elimination, in this model.
The journey to targeted therapy for malignant pDCs bearing CD123 (IL3Rα) has been arduous. Dating back 20 years, Jordan et al nicely characterized increased expression of CD123 in the leukemia stem cell compartment and hypothesized that, because of the nature of this accessible surface target, therapeutic intervention might be both clinically attractive and therapeutically feasible.4
Frankel et al investigated a novel targeted agent (DT-IL3, later known as SL- 401/tagraxofusp) in a series of patients including those with myelodysplastic syndrome (MDS), AML, and BPDCN. Although the monotherapy approach with tagraxofusp had modest clinical activity in most patients with MDS and AML in these studies, there was robust activity in patients with BPDCN (78% major response rate in the pilot study).5 This led to the pivotal phase 1/2 study, by Pemmaraju et al, for patients with BPDCN, which demonstrated 90% overall response rate in frontline setting and 67% in relapsed/refractory setting, ultimately helping to lead to drug approval specifically for patients with BPDCN.6
Given the markedly different results with CD123-tageted monotherapy with this agent in AML vs BPDCN, several questions have been raised: (1) Do these divergent results have to do with the molecular heterogeneity of AML vs BPDCN? (2) Does the quantitative or qualitative CD123 expression level matter (universally overexpressed in all BPDCN cases vs not in all cases of AML)? (3) Are there different therapeutic gradients/windows, dosing parameters, or differential organ compartment responses to consider in AML vs BPDCN? (4) Is there a treatment resistance pathway with tagraxofusp monotherapy in AML that may not be present in BPDCN? This paper from Xiao et al is very helpful in addressing these questions. This study suggests consideration of a focused targeted approach specifically in the subset of patients with pDC-AML. Based on work of Togami et al, the authors discovered a novel resistance pathway, via an acquired downregulation in diphthamide synthesis pathway (DPH1). In experimental models, the restoration of DPH1 pathway expression occurred after administration of the hypomethylating agent azacytidine in combination with tagraxofusp.7 Based on this work, Lane and Pemmaraju have initiated a novel phase 1/2 clinical trial for patients with AML and high-risk MDS in an ongoing study with 2 arms (tagraxofusp + azacytidine; tagraxofusp + azacytidine + venetoclax) (NCT03113643). It will be of great interest to analyze the results of this trial for responses in RUNX1-mutated/pDC-AML patients. Future clinical studies of pDC-AML will certainly focus on the emerging field of CD123-targeting agents. Notably, there are several other drugs approaches in active clinical trials, including novel conjugated agents, bispecific agents, and chimeric antigen receptor T-cell therapies specifically targeting CD123.
Based on this study, pDC-AML+/−RUNX1 mutations should be considered for full subcategory status in the next World Health Organization reclassification. In the latest World Health Organization 2016 reclassification by Arber et al, AML with mutated RUNX1 mutation is listed as a provisional entity.8
In summary, this study by Xiao et al confirms a new subentity of AML, pDC-AML; presents a potential novel mechanistic/mutational profile with a strong association with RUNX1 mutations; and outlines a new therapeutic avenue for investigation with CD123-targeted agents in AML. It provocatively raises the question: Is targeting pDC-AML as easy as CD 1-2-3?
Conflict-of-interest disclosure: The author reports consulting/honorarium from Celgene, Stemline, Incyte, Novartis, MustangBio, Roche Diagnostics, LFB, Pacylex, Blueprint Medicines, and Abbvie; and research funding/clinical trials support from Stemline, Novartis, Abbvie, Samus, Cellectis, Plexxikon, Daiichi-Sankyo, Affymetrix, and SagerStrong Foundation.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal