


Abstract
Microangiopathic hemolytic anemia (MAHA) with thrombocytopenia, suggests a thrombotic microangiopathy (TMA), linked with thrombus formation affecting small or larger vessels. In cancer patients, it may be directly related to the underlying malignancy (initial presentation or progressive disease), to its treatment, or a separate incidental diagnosis. It is vital to differentiate incidental thrombotic thrombocytopenia purpura or atypical hemolytic uremic syndrome in cancer patients presenting with a TMA, as they have different treatment strategies, and prompt initiation of treatment impacts outcome. In the oncology patient, widespread microvascular metastases or extensive bone marrow involvement can cause MAHA and thrombocytopenia. A disseminated intravascular coagulation (DIC) picture may be precipitated by sepsis or driven by the cancer itself. Cancer therapies may cause a TMA, either dose-dependent toxicity, or an idiosyncratic immune-mediated reaction due to drug-dependent antibodies. Many causes of TMA seen in the oncology patient do not respond to plasma exchange and, where feasible, treatment of the underlying malignancy is important in controlling both cancer-TMA or DIC driven disease. Drug-induced TMA should be considered and any putative causal agent stopped. We will discuss the differential diagnosis and treatment of MAHA in patients with cancer using clinical cases to highlight management principles.
Introduction
Microangiopathic hemolytic anemia (MAHA) refers to a subgroup of hemolytic anemia where there is fragmentation and hemolysis due to damage of erythrocytes in the small blood vessels. It is characterized by the presence of red cell fragments or schistocytes on blood film review. Further evidence of hemolysis may include a reticulocytosis, raised lactate dehydrogenase (LDH), low or absent haptoglobin, and increased unconjugated bilirubin levels. MAHA may occur in isolation due to a direct effect on red blood cells, such as trauma due to mechanical heart valves or infections (eg, malaria or march hemoglobinuria), but it is more commonly seen as part of a thrombotic microangiopathy (TMA).
The combination of MAHA and thrombocytopenia (MAHAT) clinically defines the syndrome of a TMA. Clinical features are variable and will partly depend on the underlying diagnosis.1 TMAs are characterized by endothelial cell activation and thrombus formation, leading to nonimmune hemolytic anemia, thrombocytopenia, and organ failure. Histological review reveals micro- and macrovascular thrombosis with thrombi varying in their composition depending on the cause of the TMA.
MAHAT in oncology patients may be directly related to the underlying cancer (either initial presentation or disease progression) or its treatment, or it may be a separate and incidental diagnosis. Although less common, it is vital to differentiate thrombotic thrombocytopenia purpura (TTP) as quickly as possible, as it has a distinct treatment strategy, and early initiation of treatment has a critical impact on survival.
The diagnosis of cancer-associated TMA or chemotherapy-induced TMA is crucial, as the treatment in each case is to remove the driver of the condition (either the cancer or the drug) and avoid unwarranted therapies, such as plasma exchange (PEX), which has risk of potential complications .
Here, we describe our approach to the evaluation and management of the cancer patient with MAHA using illustrative case histories. Further references where readers can find more detailed information on specific diagnoses are included. This article is intended not to provide a thorough description of each syndrome or address still-debated topics but rather to focus on practical aspects.
Case 1
A 69-year-old man with a background of congestive cardiac failure and atrial fibrillation on rivaroxaban presented with a short history of worsening fatigue. Physical examination was unremarkable. Full blood count showed hemoglobin 102 g/L; platelet count 26 × 109/L; white blood cells 5.6 × 109/L with a normal differential, and peripheral blood film examination showed a clear excess of red cell fragments with genuine thrombocytopenia and mild polychromasia. Further investigations revealed creatinine 88 μmol/L, LDH 1069 IU/L, and haptoglobin <0.1 g/L. Liver function tests showed bilirubin 26 μmol/L, alanine aminotransferase 86 IU/L, albumin 34 g/L, alkaline phosphatase 525 IU/L, coagulation screen prothrombin time (PT) 19.6 s, activated partial thromboplastin time (APTT) 31 s, and fibrinogen 1.45 g/L.
A clinical diagnosis of a TMA was made, and urgent PEX commenced pending investigations for the underlying cause. Anticoagulation was held given the thrombocytopenia. Pre-exchange ADAMTS13 activity result available the next day was normal, but a computed tomography (CT) scan of chest, abdomen, and pelvis showed a likely pancreatic primary tumor with sclerotic bone metastases, multiple hepatic metastases, and micronodularity within both lungs. PEX was stopped and oncology consult requested. He developed rapidly worsening coagulopathy and deteriorating liver function tests and was not suitable for biopsy and chemotherapy. There were no bleeding or thrombotic events. A fast-track discharge with community palliative care support was arranged, and he died on day 10 at home.
How we investigate MAHA in a cancer patient
Investigations can be considered in 2 categories: (1) those to confirm fragmentation, hemolysis, and accompanying thrombocytopenia; and (2) those that help to elucidate the underlying cause (Figure 1). TMA is defined clinically, based on standard laboratory investigations. Scoring systems, such as the PLASMIC score2 or French scoring system,3 can help to differentiate TTP from other TMAs. Definitive confirmation or exclusion of TTP can now occur in real time due to the widespread availability of commercial ADAMTS13 assays. ADAMTS13 activity levels <10% are diagnostic of TTP, while ADAMTS13 activity levels between 10% and 20% may rarely be seen in other conditions, such as severe sepsis, and require the presence of anti-ADAMTS13 immunoglobulin G autoantibodies to make a diagnosis of immune TTP.
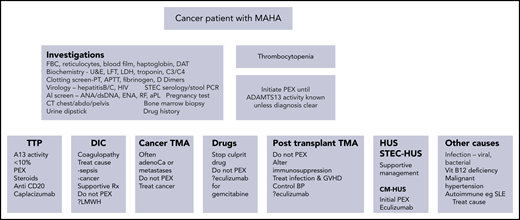
Summary of the laboratory features in patients with TMA. A differential diagnosis of the causes of a TMA are included, focusing specifically on cancer or its treatment and a summary of treatments beneficial in the individual subgroups. abdo, abdomen; adenoCa, adenocarcinoma; aPL, antiphospholipid; DAT, direct antiglobulin test; dsDNA, double-stranded DNA; ENA, extractable nuclear antigen; FBC, full blood count; GVHD, graft-versus-host disease; LMWH, low-molecular-weight heparin; PCR, polymerase chain reaction; RF, rheumatoid factor; SLE, systemic lupus erythematosus; STEC, Shiga toxin-producing Escherichia coli; U&E, urea and electrolytes; Vit, vitamin.
Summary of the laboratory features in patients with TMA. A differential diagnosis of the causes of a TMA are included, focusing specifically on cancer or its treatment and a summary of treatments beneficial in the individual subgroups. abdo, abdomen; adenoCa, adenocarcinoma; aPL, antiphospholipid; DAT, direct antiglobulin test; dsDNA, double-stranded DNA; ENA, extractable nuclear antigen; FBC, full blood count; GVHD, graft-versus-host disease; LMWH, low-molecular-weight heparin; PCR, polymerase chain reaction; RF, rheumatoid factor; SLE, systemic lupus erythematosus; STEC, Shiga toxin-producing Escherichia coli; U&E, urea and electrolytes; Vit, vitamin.
The degree of thrombocytopenia and lesser degree of renal injury in this case were in keeping with a possible diagnosis of TTP. The abnormal liver function test (LFT) results at presentation were not typical of TTP, although the patient had a history of previously mildly abnormal LFT results in an obstructive pattern, likely related to congestive cardiac failure. Similarly, the coagulation screen was not normal at presentation, but the patient was on rivaroxaban, which may cause prolongation of the PT.4 There were no specific features in the presenting symptoms to suggest an underlying diagnosis of cancer, but the LFTs and coagulation were not normal, and CT scanning was warranted. PEX was appropriate as an initial measure, pending exclusion of TTP, but was stopped when the diagnosis of a cancer-induced TMA was made.
TMA may be driven directly by the underlying cancer as is seen here. This may be the first presentation of malignancy or seen in advanced disease. Two different mechanisms are postulated to be responsible and may coexist. Systemic microvascular metastases can cause MAHAT, as vessel obstruction by tumor cells leads to fragmentation of red cells and platelet consumption in the tumor emboli (the likely mechanism in this case). MAHAT may also be due to widespread bone marrow infiltration with cancer or secondary necrosis.5-8
Most cases of cancer-associated TMA are secondary to solid organ tumors, but hematological malignancies such as lymphoma make up ∼8% of cases.9 Intravascular lymphoma is a rare but recognized cause of hemolytic anemia and neurological disorders and may present with a TTP-like picture.10,11 Gastric, lung, breast, and prostate cancers, primarily adenocarcinoma, are the most likely solid tumor diagnoses, with signet ring cell (mucin-producing) carcinoma being highlighted in several case reports.12,13
In terms of clinical features, bone pain at presentation is well described in cancer-associated TMA and is not a feature of TTP,14 likely related to cases with extensive bone marrow infiltration or secondary marrow necrosis. Respiratory symptoms (which are rare in TTP) occurred in >70% of cases of cancer-associated TMA in one case series.15 Abnormal LFT results, moderate to severe renal dysfunction, or abnormalities of the coagulation screen are also seen more frequently in cancer-associated TMA than TTP.5 Review of the blood film, an early bone marrow biopsy, and cross-sectional imaging may help expedite the underlying cancer diagnosis.16 The blood film in cancer-associated TMA may show a leucoerythroblastic picture suggesting the diagnosis.
The initial treatment of MAHAT without a clear precipitant is PEX. PEX should be undertaken as soon as the diagnosis is considered, as TTP is a hematological emergency with high untreated mortality. The benefit of PEX in TTP has been established in a randomized controlled trial,17 and PEX should be continued to clinical remission in patients with a diagnosis of TTP confirmed by ADAMTS13 testing. The requirement for ongoing PEX in other differential diagnoses will depend on the clinical response and results of additional laboratory investigations (Figure 1).
No intervention is risk-free, and potential complications of PEX include those related to central line insertion, citrate toxicity, and reactions to plasma.18 However, many of the tests to identify the etiology of the MAHAT picture should be available within 24 to 48 hours, and PEX can then be halted if another diagnosis is identified for which PEX has no benefit. If TTP is confirmed by demonstrating severe ADAMTS13 deficiency, then further targeted treatment is required with immunosuppression (steroids and anti-CD20 therapy), plus adjunctive caplacizumab (an anti-von Willebrand factor [anti-VWF] nanobody that blocks formation of VWF-platelet microthrombi).19
Making the diagnosis of a cancer-associated TMA is crucial, as there is no beneficial role for PEX, steroids, or other immunosuppression that is used in TTP. Platelet transfusions may be given for severe thrombocytopenia in a case of cancer-associated TMA, based on the clinical picture and following usual platelet transfusion thresholds, unlike in TTP, where platelet transfusions are usually avoided because of risk of exacerbating microthrombotic complications. Treatment with chemotherapy has been associated with improved survival in cancer-associated TMA,9 but many patients have an extremely limited prognosis, with nearly a 50% mortality rate within 1 month of diagnosis,5,9,20 as highlighted in our case. His rapid death was probably not preventable, given the advanced stage of the cancer at presentation.
Case 2
A 59-year-old man presented with a subglottic mass requiring a laryngopharyngectomy for squamous cell carcinoma, followed by chemotherapy and radiotherapy. Four months later, he presented with vomiting and retrosternal chest pain. Blood tests showed hemoglobin 81 g/L, platelets 42 × 109/L, reticulocytes 4.5%, and normal renal function. Schistocytes were noted on the blood film. He deteriorated rapidly, requiring intubation and ventilation and inotrope support. Further investigations revealed LDH 310 IU/L, APTT 49 s, PT 10.9 s, and fibrinogen 4.4 g/L. D-dimers were 23 900 μg/L fibrinogen equivalent units, C-reactive protein (CRP) 207 mg/L, and alkaline phosphatase 663 IU/L. ADAMTS13 activity was normal, and the patient continued with supportive care. His coagulation screen worsened to a PT of 17 s, APTT 55 s, and fibrinogen 0.5g/L. He was bleeding from the endotracheal tube and urinary catheter, and bruises were noted on his arms, legs, and torso. A CT scan of the chest, abdomen, and pelvis revealed bilateral renal cortical infarcts, bilateral consolidation, and a large right-sided pleural effusion. A mass suspicious of metastatic cancer was noted in the upper lobe of his right lung. An occipital infarct was noted on the CT head. Aspiration of the pleural effusion, sent for culture, identified Streptococci milleri and Candida albicans. An echocardiogram was normal.
In this case, the initial picture based on routine blood counts and the acute clinical scenario suggested a TMA. However, the LDH was only mildly elevated, the CRP was high, and the coagulation screen was deranged and deteriorated.
Disseminated intravascular coagulation (DIC) is a clinicopathological syndrome that can be precipitated in cancer patients by sepsis or driven by the disease itself. This patient had metastatic cancer and 2 confirmed infective agents, likely causing the DIC picture, with evidence of bleeding and thrombosis.
Thrombocytopenia is the initial and most sensitive sign of DIC, occurring in >90% of cases, and half of cases have platelet counts <50 × 109/L.21 The falling platelet count is associated with an increase in thrombin formation and fibrinolytic activity, resulting in raised D-dimers. There is variability in the results of the coagulation screen (PT and/or APTT). Clotting times are prolonged in 60% to 70% cases but may be normal or even shortened. Fibrinogen levels may be reduced but are not usually below the normal reference range unless the DIC is very severe.21 This case demonstrates how fibrinogen levels were in the normal laboratory range in the earlier stages of DIC and that it is the progressive changes in these parameters that is more helpful in confirming the diagnosis of DIC. Use of scoring systems for DIC improves the accuracy of diagnosis. The International Society of Thrombosis and Haemostasis scoring system demonstrates >90% sensitivity and specificity and is associated with clinical outcome (ie, increased mortality).21
Despite prolongation of coagulation parameters and thrombocytopenia, in cancer patients, the principal clinical feature may be thrombosis rather than hemorrhage, or, as in this case, both may occur simultaneously. Although not present in this case, severe sepsis-related DIC can be associated with reduced ADAMTS13 activity with increased large VWF multimers and related to the risk of acute kidney injury.22 ADAMTS13 is synthesized in the liver; thus, any degree of hepatic failure may result in reduced ADAMTS13 activity. However, the levels of ADAMTS13 activity are not typically <10 IU/dL, as is the case in acute TTP.
Certain cancers are more likely to provoke DIC, particularly adenocarcinomas of the gastrointestinal tract (frequently signet ring cell type), pancreas, breast, prostate, or lung. Mucinous tumors may secrete enzymes that can activate factor X.5 DIC occurs in patients with acute promyelocytic leukemia, caused by release of procoagulants by abnormal promyelocytes23 and also in acute monocytic leukemia.
Treatment of cancer-associated DIC is primarily of the underlying driver of the condition, be it the malignancy or sepsis. Patients may require blood product support if bleeding or requiring a procedure, aiming for a PT and APTT <1.5 upper limit of normal, fibrinogen >1.5 g/L, and platelets >50 × 109/L. Adjunctive vitamin K may be used, especially if LFT results are abnormal, affecting coagulation factor production. Concurrently, initiation of low-molecular-weight heparin may be required, at least at thromboprophylactic levels, if the phenotype is predominantly thrombotic
Management in this case was therefore complex. There was no further therapy available for his cancer. Appropriate antibiotics based on sensitivities and antifungal therapy were initiated. Vitamin K was started 10 mg IV daily for 3 days, and 10 mL/kg fresh frozen plasma with cryoprecipitate was given. Further blood products were administered to improve the coagulation parameters, stop bleeding, and allow initiation of low-molecular-weight heparin, initially at prophylactic intensity and incremented based on laboratory tests and clinical findings. This case highlights a number of potential precipitants causing MAHA with a DIC picture, but treatments are supportive.
Case 3
A 50-year-old man was diagnosed with myeloma, having previously been completely fit and well. He received 4 cycles of cyclophosphamide, dexamethasone, and carfilzomib and was continued on carfilzomib maintenance therapy. Fifteen months later, he presented with a 1-week history of nausea and reduced urine output, having had a normal full blood count and renal function the preceding week. On admission, hemoglobin was 89 g/L, platelets 18 × 109/L, reticulocytes 1.05%, haptoglobin <0.1 g/L, LDH 3070 IU, CRP 24, urea 35.8 mmol/L, and creatinine 748 μmol/L; the coagulation screen was normal, but schistocytes was evident on a blood film. PEX and hemofiltration were initiated. ADAMTS13 activity was normal. His blood pressure was increased to 160/90. Carfilzomib was stopped. He received 5 daily PEX procedures with rapid resolution of his MAHA and normalization of the platelet count. He required ongoing renal replacement therapy on discharge and triple antihypertensive therapy. Four months later, his hematology parameters remained normal, creatinine 119 umol/L and eGFR 58mls/min off renal replacement therapy, but continued on antihypertensive therapy. His paraprotein was undetectable.
Proteasome inhibitors (PIs) are now known to be associated with a TMA,24,25 at a rate greater than that of complement-mediated hemolytic-uremic syndrome (HUS), which has an incidence of 1 or 2 per million. The clinical picture is comparable to HUS with more severe renal impairment than is usually seen in other TMAs.
PI-induced TMA can be a challenging diagnosis in patients with myeloma. The differential diagnoses for worsening thrombocytopenia include disease progression or myeloma therapy, while renal impairment is common in the myeloma patient with a wide differential. In these patients, an acute and unexplained thrombocytopenia, hemolysis, and renal failure (with hypertension), while the disease itself is under control, should lead clinicians to consider this complication. Monitoring of blood pressure during PI therapy is important, with prompt withdrawal of the medication if there is evidence of emerging TMA, given the timing of this complication cannot be predicted.
Currently, there are 3 FDA-approved PIs: bortezomib, carfilzomib, and ixazomib. Bortezomib, the first PI that was approved, is currently indicated for the treatment of patients with multiple myeloma and mantle cell lymphoma. Carfilzomib and ixazomib are indicated for the treatment of relapsed/refractory myeloma. The prevalence of TMA with carfilzomib is much higher than with bortezomib.25 Isolated microangiopathic hemolytic anemia, without all the features of a full-blown TMA, was observed at very high rates in carfilzomib-treated patients in a recent case series where it occurred in 16 out of 24 patients (67%).26 In this single-center cohort, 11 out of 16 patients had mild hemolysis with haptoglobin <0.1 g/L but no transfusion requirement; 5 out of 16 required transfusion, and 1 patient had a severe and subsequently fatal TMA.26 Cases of TMA have also been reported with the newer generation of PIs such as ixazomib.27
The most important principles of management of PI-induced TMA are cessation of the culprit agent, blood pressure control, and renal replacement therapy where required. The relative role of PEX and/or complement inhibition as part of the supportive management strategy in proteasome-induced TMA is not yet clear. PEX may be helpful in improving the hematology parameters, as is seen in complement-mediated HUS (CM-HUS, previously known as atypical HUS).28
CM-HUS itself is initially a diagnosis of exclusion. The diagnosis is subsequently confirmed by the presence of mutations in the alternative complement pathway affecting its regulation, which are found in approximately two-thirds of cases.29 Definitive treatment of CM-HUS, however, is with complement inhibition (eculizumab), and the earlier anticomplement therapy is started, the better the longer-term renal outcome.30
Guidelines published by one US group on the management of PI-induced TMA argue against the use of PEX and for initial empiric treatment with eculizumab.31 One group found C5b-C9 deposition on endothelial cells in culture exposed to plasma from patients with acute carfilzomib-induced TMA, potentially allowing identification of patients who could benefit from complement blockade.32 However, further data are needed before any definitive recommendation about the role of these therapies in PI-induced TMA can be made.
Drug-associated TMA
Drugs are a rare but increasingly important cause of MAHA, usually in association with thrombocytopenia, and may cause TMA either by cumulative dose-dependent toxicity or an idiosyncratic reaction after development of drug-dependent antibodies.33
Extremely rarely, a drug may be associated with true anti-ADAMTS13 antibody-mediated TTP. This has been described for the antiplatelet agent ticlopidine34 and more recently as a possible association of the immune checkpoint inhibitors that are used to treat metastatic melanoma and other cancers and the immunomodulatory drug lenalidomide, which is used to treat multiple myeloma.
Three cases have been described in which patients developed immune TTP with ADAMTS13 inhibitors during checkpoint inhibitor therapy (1 case with ipilimumab and 2 cases with dual checkpoint inhibitor therapy [ipilimumab plus nivolumab]) that responded to PEX, steroids, and rituximab and cessation of the drug.35-37 TTP with severe ADAMTS13 deficiency has been described in 5 cases of patients treated with lenalidomide, 4 of whom had detectable anti-ADAMTS13 antibodies, who were successfully managed with PEX, immunosuppression, and discontinuation of the drug.38-40 While there is no direct evidence for a role of the drug as a cause of the anti-ADAMTS13 antibodies and hence ADADMTS3 deficiency, a wide variety of other immune-related adverse events have been reported with both checkpoint inhibitors and lenalidomide.41
However, in the vast majority of cases of drug-associated TMA, the mechanism is not ADAMTS13 mediated, and clinical presentation involves primarily renal impairment in conjunction with MAHAT. Sudden onset of symptoms that recur with repeated administration of a drug should prompt consideration of an immune mechanism with a drug-dependent antibody. However, if there is slowly progressive kidney injury with MAHAT, then dose-dependent toxicity is more likely.5
Some anticancer agents have long been associated with TMA. Probably the best described is gemcitabine, which causes cumulative dose-dependent toxicity, with damage predominantly to the renal endothelium. In a large national retrospective cohort of 120 French patients with gemcitabine TMA, diagnosis occurred after a median of 210 days of treatment and a cumulative dose of 12 941 mg/m2.42 Ninety-five percent had MAHA, and three-quarters were thrombocytopenic. Almost all patients (97%) had an acute kidney injury, with dialysis required in 28%.42
TMAs occurring after vascular endothelial growth factor (VEGF) inhibitors (eg, bevacizumab) are a relatively common complication in oncology.43 Proteinuria is the earliest feature, followed by hypertension and renal failure. Cytopenias are seen with MAHA. The mechanism is dose-dependent toxicity and renal biopsy demonstrates microthrombi limited to glomerular capillaries with glomerular basal membrane alterations due to VEGF inhibition in podocytes.43 Treatment consists of stopping the culprit agent, and the outcome is usually favorable.
Oxaliplatin therapy is associated with the formation of drug-dependent antibodies against platelets and erythrocytes, and an acute onset immune-mediated TMA is well recognized.33,44,45 A list of other drugs used in the setting of cancer that have been associated with MAHAT, with potential mechanisms and management strategies, is given in Table 1.
Drugs used in the setting of cancer and associated with MAHA
| Cancer therapy | Potential mechanism of TMA and management |
|---|---|
| Checkpoint inhibitors (eg, ipilimumab) | ADAMTS13 deficiency (ADAMTS13 inhibitor present); responds to PEX |
| Lenalidomide | ADAMTS13 deficiency (anti-ADAMTS13 Ab in 4/5 cases); responds to PEX |
| Gemcitabine | Dose-dependent endothelial damage, predominantly renal glomerular arterioles/capillaries; may respond to complement inhibition |
| Mitomycin C | Dose-dependent toxicity, microthrombi in glomerular arterioles/capillaries; may respond to complement inhibition |
| VEGF inhibitors (eg, bevacizumab, aflibercept) | Dose-dependent toxicity; hypertension; microthrombi limited to glomerular capillaries |
| Proteosome inhibitors (eg, bortezomib, carfilzomib) | Renal impairment and hypertension with TMA; favorable response to stopping culprit drug; may respond to complement inhibition; role of PEX? |
| Pentostatin | Dose-dependent toxicity at high doses |
| EGFR inhibitor cetuximab | Renal TMA with nephrotic syndrome |
| Calcineurin inhibitors (eg, ciclosporin, tacrolimus) | TMA primarily affects glomerular arterioles; reducing dose or stopping drug can improve or reverse the TMA |
| mTOR inhibitors (eg, sirolimus, everolimus) | Renal TMA; can occur in patients on calcineurin inhibitor–free regimen |
| Platinum-based agents (eg, oxaliplatin) | Drug-dependent antibodies against platelets and red cells |
| Hormone therapies (eg, tamoxifen) | Precipitation of congenital TTP/association with immune TTP; close ADAMTS13 monitoring required if history of TTP |
| Cancer therapy | Potential mechanism of TMA and management |
|---|---|
| Checkpoint inhibitors (eg, ipilimumab) | ADAMTS13 deficiency (ADAMTS13 inhibitor present); responds to PEX |
| Lenalidomide | ADAMTS13 deficiency (anti-ADAMTS13 Ab in 4/5 cases); responds to PEX |
| Gemcitabine | Dose-dependent endothelial damage, predominantly renal glomerular arterioles/capillaries; may respond to complement inhibition |
| Mitomycin C | Dose-dependent toxicity, microthrombi in glomerular arterioles/capillaries; may respond to complement inhibition |
| VEGF inhibitors (eg, bevacizumab, aflibercept) | Dose-dependent toxicity; hypertension; microthrombi limited to glomerular capillaries |
| Proteosome inhibitors (eg, bortezomib, carfilzomib) | Renal impairment and hypertension with TMA; favorable response to stopping culprit drug; may respond to complement inhibition; role of PEX? |
| Pentostatin | Dose-dependent toxicity at high doses |
| EGFR inhibitor cetuximab | Renal TMA with nephrotic syndrome |
| Calcineurin inhibitors (eg, ciclosporin, tacrolimus) | TMA primarily affects glomerular arterioles; reducing dose or stopping drug can improve or reverse the TMA |
| mTOR inhibitors (eg, sirolimus, everolimus) | Renal TMA; can occur in patients on calcineurin inhibitor–free regimen |
| Platinum-based agents (eg, oxaliplatin) | Drug-dependent antibodies against platelets and red cells |
| Hormone therapies (eg, tamoxifen) | Precipitation of congenital TTP/association with immune TTP; close ADAMTS13 monitoring required if history of TTP |
Ab, antibody; EGFR, endothelial growth factor receptor; mTOR, mammalian target of rapamycin.
When evaluating oncology patients with MAHA, their anticancer therapy and other drugs should be considered as a possible cause and any potential culprit agent stopped immediately. This may be difficult in those therapies resulting in TMAs following a cumulative drug dose. The differential between cancer-associated TMA, chemotherapy-associated TMA, and CM-HUS can be challenging, as all are diagnoses of exclusion and none are associated with severe ADAMTS13 deficiency.
There is a very limited role for PEX in drug-associated TMA, as only a small proportion of cases (those related to ticlopidine and potentially the immune checkpoint inhibitors or the immune modulatory drug lenalidomide) have antibodies against ADAMTS13 .
There are an increasing number of case reports of anticomplement therapy being used in the management of drug-associated TMA, particularly where the mechanism is dose-dependent toxicity, as seen with gemcitabine and mitomycin C, given the similarity of the presentation with CM-HUS.42,46-49 Some groups have identified mutations or polymorphisms in complement regulatory genes in patients who respond, suggesting a role for complement dysregulation in the underlying pathophysiology.50 Further data are needed before any definitive recommendation about the role of complement inhibition in subgroups of drug-induced TMA can be made.
TA-TMA
Transplant-associated TMA (TA-TMA) may affect allogeneic haemopoietic cell transplant (allo-HCT) or solid-organ transplant patients. The following discussion centers around our management approach to TA-TMA as a complication of allo-HCT. TA-TMA is associated with a high mortality, no definitive diagnostic criteria, and therapy limited to supportive care, with no beneficial role for PEX.51 It presents with MAHAT, and renal dysfunction, hypertension, and neurological features such as seizures are common organ-related symptoms.52 Diagnosis of TA-TMA may be difficult, especially in the setting of conditioning-related cytopenias.53
There are a number of factors that contribute to and may have an additive effect resulting in endothelia cell injury. These include the conditioning therapy, HLA-associated mismatches (particularly in unrelated donors), and high therapeutic levels of calcineurin inhibitors used to prevent graft rejection. Additional infections, especially viruses such as adenovirus or cytomegalovirus, need to be excluded and treated. Proteinuria, raised LDH, and hypertension have been used as prognostic factors and associated with increased mortality.52
PEX has not been shown to demonstrate a positive benefit in TA-TMA, but reduction or using an alternative immunosuppressive regimen to prevent graft-versus-host disease and treatment of coexisting infections, meticulous blood pressure control, and general supportive therapy. More recently, as serological, cellular, and genetic evidence of complement activation has been demonstrated in TA-TMA,54,55 the use of eculizumab has been increasingly reported.56 Newer agents have also been used that target nitric oxide pathways.51 However, it is becoming clear that not all allo-HCT TA-TMA is complement driven; thus, identifying a reliable biomarker that can ascertain these cases will be helpful in directing therapy.53,57
In contrast, recurrent TMA after renal transplant may represent recurrence of CM-HUS (atypical HUS) or related to calcineurin inhibitors and should be managed accordingly.
Other differential diagnoses
Infections may present with MAHAT and are particularly relevant in patients who are immunosuppressed because of chemotherapy. TTP with severe ADAMTS13 deficiency may be a presentation of HIV. Many other infections may present with a MAHA and low platelets, such as adenovirus, cytomegalovirus, or even tuberculosis, and differential diagnosis requires not only a careful history but also detailed microbiological and virology investigations. Treatment requires the appropriate antimicrobial therapy.58
Severe vitamin B12 deficiency may cause anemia with significant red cell fragmentation, and LDH levels are often markedly elevated. The condition may mimic a TMA with severe thrombocytopenia, but the issue is ineffective hematopoiesis with low reticulocytes, and the thrombocytopenia is due to a failure of bone marrow production. The presentation may be difficult to distinguish from TTP, and PEX has even been initiated in some cases.59 Other causes of a MAHA include malignant hypertension and autoimmune conditions such as vasculitis or lupus nephritis. Management of these other causes of MAHAT involves treating the underlying condition (eg, blood pressure control for malignant hypertension), and PEX should be discontinued.
In conclusion, the differential diagnosis of MAHAT in patients with cancer is wide. Early exclusion of TTP is essential, but it is the subsequent diagnosis that drives appropriate management. PEX is not effective in many of the causes of TMA seen in cancer patients, and, where feasible, treatment of the underlying malignancy is important in controlling both cancer-associated TMA and DIC driven by the disease. The possibility of drug-associated TMA should be considered and any potential causative medication stopped. There is emerging evidence for the potential role of complement inhibition in selected cases of drug-associated and TA-TMA.
Authorship
Contribution: M.R.T. and M.S. wrote the paper and reviewed the final version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: M. Scully, University College London Hospitals, 250 Euston Rd, London, nw1 2pg, United Kingdom; e-mail: m.scully@ucl.ac.uk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal