

Key Points
In a series of 29 856 CLL patients, the incidence of BcR stereotypy peaked at 41%.
Higher-order relations exist between stereotyped subsets, particularly for those from U-CLL, for which satellite subsets were identified.

Abstract
Chronic lymphocytic leukemia (CLL) is characterized by the existence of subsets of patients with (quasi)identical, stereotyped B-cell receptor (BcR) immunoglobulins. Patients in certain major stereotyped subsets often display remarkably consistent clinicobiological profiles, suggesting that the study of BcR immunoglobulin stereotypy in CLL has important implications for understanding disease pathophysiology and refining clinical decision-making. Nevertheless, several issues remain open, especially pertaining to the actual frequency of BcR immunoglobulin stereotypy and major subsets, as well as the existence of higher-order connections between individual subsets. To address these issues, we investigated clonotypic IGHV-IGHD-IGHJ gene rearrangements in a series of 29 856 patients with CLL, by far the largest series worldwide. We report that the stereotyped fraction of CLL peaks at 41% of the entire cohort and that all 19 previously identified major subsets retained their relative size and ranking, while 10 new ones emerged; overall, major stereotyped subsets had a cumulative frequency of 13.5%. Higher-level relationships were evident between subsets, particularly for major stereotyped subsets with unmutated IGHV genes (U-CLL), for which close relations with other subsets, termed “satellites,” were identified. Satellite subsets accounted for 3% of the entire cohort. These results confirm our previous notion that major subsets can be robustly identified and are consistent in relative size, hence representing distinct disease variants amenable to compartmentalized research with the potential of overcoming the pronounced heterogeneity of CLL. Furthermore, the existence of satellite subsets reveals a novel aspect of repertoire restriction with implications for refined molecular classification of CLL.
Introduction
Antigen interactions mediated by the B-cell receptor (BcR) immunoglobulin are critical for the survival and proliferation of chronic lymphocytic leukemia (CLL) malignant cells.1,2
From a clinical perspective, this notion was corroborated by the therapeutic efficacy of pharmacological agents inhibiting kinases downstream the BcR signaling pathway, even in relapsed/refractory CLL patients.3-5 From a biological standpoint, support for the role of antigenic interactions in CLL was provided by pivotal studies from the 1990s reporting significant skewing in immunoglobulin gene usage, that was different from that of normal B cells.6 Soon thereafter, the somatic hypermutation (SHM) status of the clonotypic rearranged immunoglobulin heavy variable (IGHV) genes was shown to strongly correlate with patient outcome,7-9 a finding of seminal importance for both understanding and managing CLL.10
The relevance of the BcR immunoglobulin in CLL was further reinforced through the identification of subsets of patients expressing stereotyped BcR immunoglobulin.11-15 In our previous study of 7424 patients, we reported that ∼30% of all CLL were assigned to stereotyped subsets, each defined by a distinctive amino acid pattern within the variable heavy complementarity determining region 3 (VH CDR3) of the clonotypic BcR immunoglobulin.15 Moreover, we identified 19 sizeable subsets that were defined as major, collectively accounting for 12.4% of all CLL.15
Accumulating evidence indicates that patients assigned to the same stereotyped subset exhibit consistent biological background as reflected in similar profiles of antigen reactivity,16-20 genomic aberrations,21-23 gene expression,24,25 epigenetic modifications,26-28 Toll-like receptor signaling,29,30 "classic,”31 and cell-autonomous32,33 BcR signaling, among others. Moreover, BcR immunoglobulin stereotypy defines subgroups with consistent clinical presentation and outcome, particularly exemplified by: (1) subset #2 that is associated with aggressive clinical course regardless of the SHM status and appears unresponsive to chemoimmunotherapy,34,35 and (2) subset #8 that displays the highest risk for Richter transformation among all CLL. Hence, information regarding subset membership can contribute to refined risk stratification of CLL patients,36-42 at least for certain subsets.
Altogether, these findings support the notion that the study of BcR immunoglobulin stereotypy in CLL has important implications for both understanding disease pathophysiology and refining clinical decision-making.34,35,38,41,42 That said, several issues remain open, especially pertaining to: (1) the exact frequency of BcR immunoglobulin stereotypy and of the major subsets; (2) its relation to IGHV gene usage and SHM status; and (3) the existence of high-order relationships between subsets.
To this end, we undertook an in-depth study of BcR immunoglobulin stereotypy in a cohort of 29 856 patients with CLL, 4 times larger compared with our previous study.15 We report a significant increase in the relative size of the stereotyped fraction, which now exceeds 41% of all CLL. Stereotyped subsets previously designated as major retained their relative size, while 10 novel major subsets emerged. Finally, high-order relationships were detected between subsets, with certain major U-CLL subsets displaying several closely related “satellite” subsets, often of considerable size. This finding argues for a similar pathophysiology and clonal behavior of these subsets, with implications for refined risk stratification and improved clinical decision-making.
Methods
Patient group
Included in the study were 29 856 patients with CLL from 45 collaborating Institutes in Europe, 3 in the United States, and 1 in Asia. A subgroup of 7424 patients included in the present cohort have been reported in our previous study.15 Furthermore, cases from the cohort contributed by the German CLL Study Group (GCLLSG) have been investigated for membership to stereotyped subsets #1, #2, #4, and #8 in the context of clinicobiological association studies.35 Detailed information regarding the geography and the contribution of each Institute is given in supplemental Table 1 (available on the Blood Web site). All cases met the diagnostic criteria of the International Workshop on Chronic Lymphocytic Leukemia (iwCLL).43 The study was approved by the local ethics review committee of each participating Institute, and conducted with informed consent.
PCR amplification and sequence analysis
Polymerase chain reaction (PCR) amplification of IGHV-IGHD-IGHJ gene rearrangements was performed prior to the application of any treatment on either genomic DNA (gDNA) or complementary DNA (cDNA), as described.15 PCR products were gel-excised, purified and subjected to bidirectional direct sequencing.44 Sequence data were annotated with the IMGT/HighV-QUEST tool.45 Only in-frame, productive rearrangements were subjected to further analysis.
Metadata analysis
IMGT/HighV-QUEST output data were processed with available in-house algorithms. Information regarding IGH gene repertoires, SHM patterns, and VH CDR3 characteristics was extracted, as described.15
Subset assignment of IGH sequences
Stereotyped BcR IGHV-IGHD-IGHJ gene rearrangements were identified using our established bioinformatics method.15,46 Briefly, the applied clustering criteria were: (1) utilization of IGHV genes from the same phylogenetic clan, (2) at least 50% amino acid identity and 70% similarity within the VH CDR3, (3) identical VH CDR3 length and, (4) identical offset of the shared amino acid pattern. Common sequence patterns between sequences belonging to different clusters led to their grouping in clusters at progressively higher levels of hierarchy describing more distant, thus more relaxed, sequence relationships with more widely shared sequence patterns.
A subset was defined as major if it represented at least 0.2% of the cohort, that is, when containing at least 60 cases; a similar cutoff (0.26%) had been used for identifying major subsets in our previous study.15
Sequence relatives of major subsets, that is, subsets with similar immunogenetic features, hereafter termed satellites, were identified using the following parameters: (1) utilization of phylogenetically related IGHV genes, (2) maximum VH CDR3 length difference of 2 aa, and, (3) presence of the “subset-specific” VH CDR3 sequence motifs. The analysis was performed individually for each major subset.
Subset nomenclature
To avoid potential confusion, the original subset names were retained for stereotyped subsets reported in previous studies.11,12,14 For novel subsets, a new nomenclature was adopted, whereby subset names consisted of 4 bits, separated by vertical lines: (1) IGHV gene(s) used, (2) IGHJ gene used, (3) VH CDR3 amino acid length, (4) a numerical character in serial order (in the case of subsets with identical IGHV gene and CDR3 length, for example, 2 distinct novel subsets utilizing the IGHV3-23 and IGHJ4 genes and carrying VH CDR3s of 13 aa would be termed V3-23|J4|13|1 and V3-23|J4|13|2). Detailed information is provided in supplemental Methods.
Data visualization tools
VH CDR3 amino acid patterns of different subsets were visualized using WebLogo (http://weblogo.berkeley.edu/). Each logo consists of stacks of symbols, one stack for each position of the sequence. Amino acid position numbering is according to the IMGT numbering for the V domain.
Statistical analysis
Descriptive statistics for discrete variables included counts and frequency distributions. For quantitative variables, statistical measures included the mean, median, and min–max values. The significance of bivariate/multivariate relationships between variables was assessed using the χ2 and unpaired Student t tests. For all comparisons a significance level of P = .05 was set. All statistical analyses were performed with SPSS V-22 (https://www.ibm.com) and R V-3.6.1 (www.r-project.org).
Results
IGH gene repertoire and somatic hypermutation analysis
Overall, 30 413 IGHV-IGHD-IGHJ gene rearrangements were amplified from 29 856 CLL patients; in 29 319 cases (98.2%), a single productive rearrangement was identified, whereas 2 productive rearrangements were detected in 517 cases (1.7%); finally, 20 cases (0.1%) carried 3 distinct IGH rearrangements. The IGH gene repertoires were in line with our previous study15 (Figure 1; supplemental Results; supplemental Tables 2 and 3).

The IGHV gene repertoire of stereotyped CLL is clearly distinct from the general cohort. Cases expressing the IGHV1-69 and IGHV3-21 genes were significantly overexpressed in the stereotyped fraction of CLL compared with the heterogeneous group (16.4% vs 12.5% and 7.9% vs 4.8%, respectively). On the other hand, cases carrying the IGHV3-23 showed the opposite trend (4.6% in the stereotyped fraction vs 7.6% in the general cohort). Finally, the frequency of IGHV4-34 expressing cases was high in both groups (8.8% vs 8.7%).
The IGHV gene repertoire of stereotyped CLL is clearly distinct from the general cohort. Cases expressing the IGHV1-69 and IGHV3-21 genes were significantly overexpressed in the stereotyped fraction of CLL compared with the heterogeneous group (16.4% vs 12.5% and 7.9% vs 4.8%, respectively). On the other hand, cases carrying the IGHV3-23 showed the opposite trend (4.6% in the stereotyped fraction vs 7.6% in the general cohort). Finally, the frequency of IGHV4-34 expressing cases was high in both groups (8.8% vs 8.7%).
The classification into the unmutated CLL (U-CLL) and mutated CLL (M-CLL) subgroups was based on the 98% germline identity (GI) cutoff value. Among 29 319 cases carrying a single productive rearrangement, 15 859 (54.1%) were classified as M-CLL (GI < 98%), and 13 460 (45.9%) as U-CLL (GI ≥ 98%) (Table 1). More information can be found in the supplemental Results.
Somatic hypermutation status at cohort level
| Mutational groups | BcR immunoglobulin rearrangements | |
|---|---|---|
| No. | % | |
| Truly unmutated, GI = 100% | 9.585 | 32.7 |
| Unmutated, 98% ≤ GI <99.9% | 3.875 | 13.2 |
| Mutated, GI <98% | 15.859 | 54.1 |
| Mutational groups | BcR immunoglobulin rearrangements | |
|---|---|---|
| No. | % | |
| Truly unmutated, GI = 100% | 9.585 | 32.7 |
| Unmutated, 98% ≤ GI <99.9% | 3.875 | 13.2 |
| Mutated, GI <98% | 15.859 | 54.1 |
The stereotyped fraction accounts for a large percentage of all CLL
In total, 12 541 of 30 413 sequences, representing 41.2% of the entire cohort, were assigned to 5157 distinct ground level clusters, including 2 to 160 sequences each. In many cases, the existence of multiple shared patterns within VH CDR3 sequences led to their concurrent assignment in different ground-level subsets. This overlap enabled the discovery of subsets at 4 successive hierarchically higher levels, characterized by more broadly shared sequence patterns and, hence, greater individual subset size. When considering all clustering levels, we identified 2195 distinct stereotyped subsets ranging in size from 2 to 764 sequences each. Interestingly, random set simulations indicated that the size of the cohort correlated with the number of stereotyped subsets but not their relative frequency (supplemental Results; supplemental Figure 1).
Major stereotyped subsets
Twenty-nine major subsets with an individual frequency of at least 0.2% of the entire cohort were identified, cumulatively accounting for 4099 of 30 413 (13.5%) of all CLL and for almost one-third (32.7%) of the stereotyped fraction. These included the 19 previously identified major subsets and 10 novel ones. Subset #2 (comprising cases from both M-CLL and U-CLL)34 was the largest (764 sequences, 2.5%), followed by subsets #1 (U-CLL, 651 sequences, 2.1%) and #4 (M-CLL, 265 sequences, 0.9%). Subset #148B (M-CLL, 152 sequences, 0.5%) was the largest of the novel major subsets (Figure 2). Most major subsets concerned U-CLL (18 of 29 subsets, 62%). Detailed information for all major stereotyped subsets is provided in supplemental Tables 4 and 5, whereas a list of all minor subsets is given in supplemental Table 6.
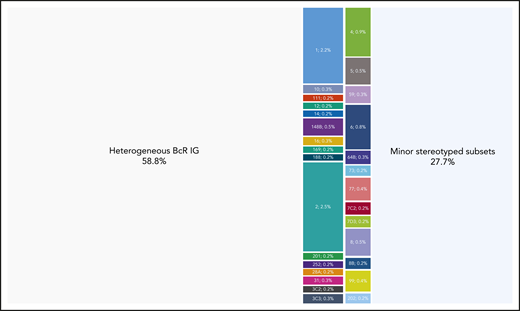
Cases assigned to major stereotyped subsets represent a significant portion of CLL. Twenty-nine different subsets were identified in the present study containing a minimum of 60 cases (0.2% of the cohort) and were defined as major. The relative size of each major subset is indicated in the graph. Their basic immunogenetic information is given in supplemental Table 4; all sequences assigned to each major subset are listed in supplemental Table 5. Altogether, major subsets comprised in total 4098 of 30 413 rearrangements that corresponds to the 13.5% of the cohort.
Cases assigned to major stereotyped subsets represent a significant portion of CLL. Twenty-nine different subsets were identified in the present study containing a minimum of 60 cases (0.2% of the cohort) and were defined as major. The relative size of each major subset is indicated in the graph. Their basic immunogenetic information is given in supplemental Table 4; all sequences assigned to each major subset are listed in supplemental Table 5. Altogether, major subsets comprised in total 4098 of 30 413 rearrangements that corresponds to the 13.5% of the cohort.
IGHV gene usage in stereotyped CLL subsets
A single IGHV gene clearly predominated in the stereotyped IGHV-IGHD-IGHJ gene rearrangements of most major subsets (23 of 29, 79.3%). Within this category of “single dominant IGHV” subsets, IGHV1-69 predominated (6 of 23 subsets, 26%), followed by the IGHV4-34, IGHV4-39 and IGHV3-48 genes (each found in 3 of 23 subsets, 13%). Single dominant major subsets exhibited concordant SHM status.
Three of the remaining 6 major subsets (#12, #59, and #77)15 included rearrangements utilizing 2 different IGHV genes with comparable frequencies. In all 3 cases, the respective IGHV genes belonged to the same phylogenetic clan and, thus, exhibited high sequence similarity: IGHV1-2 and IGHV1-46 in subset #12 (U-CLL), IGHV1-58 and IGHV1-69 in subset #59 (U-CLL), and, finally, IGHV4-4 and IGHV4-59 in subset #77 (M-CLL), respectively (supplemental Figure 2).
The remaining 3 major subsets included cases expressing several different, yet phylogenetically related IGHV genes; these included the well-established subset #1, its closely related subset #99 (both exhibiting rearrangements of clan I IGHV genes), and subset #202 (clan III IGHV genes). All 3 patterns of IGHV gene usage (ie, single dominant IGHV, “2 IGHV/same clan,” “multiple IGHV/same clan”) were also evident in minor subsets (supplemental Table 7).
Different BcR immunoglobulin stereotypy profiles in U-CLL and M-CLL
The incidence of stereotyped BcR immunoglobulin differed significantly between U-CLL vs M-CLL. Specifically, 53% of U-CLL BcR IGs were assigned to stereotyped subsets, compared with 34% in M-CLL (P < .05; Figure 3). In our previous study,15 the respective values were 41% for U-CLL and 22% for M-CLL: hence, we have now uncovered a significant increase (P < .05) in the frequency of BcR immunoglobulin stereotypy in both mutational subgroups.

Frequency of BcR immunoglobulin stereotypy in the CLL mutational subgroups. The majority of cases carrying BcR immunoglobulin sequences belonging to U-CLL were assigned to stereotyped subsets compared with approximately one-third of cases from M-CLL.
Frequency of BcR immunoglobulin stereotypy in the CLL mutational subgroups. The majority of cases carrying BcR immunoglobulin sequences belonging to U-CLL were assigned to stereotyped subsets compared with approximately one-third of cases from M-CLL.
Most major U-CLL subsets exhibited high conservation across the entire VH CDR3. Among major subsets, the most widely conserved motif was identified in subset #10 (supplemental Figure 3A), where the subset-defining motif comprised 20 of 22 VH CDR3 amino acids. Such remarkable conservation was also evident in minor subsets, for example, minor subset #19A (U-CLL) where the shared motif comprised 17 of 19 VH CDR3 amino acids (supplemental Figure 4A).
In contrast, M-CLL stereotyped subsets were often characterized by relatively degenerate sequence motifs with only few highly restricted (“landmark”) amino acid residues. Indicatively, in major subset #148B the conserved residues were mostly located at either the 5′ (IGHV-encoded) or the 3′ (IGHJ-encoded) part of the VH CDR3. Most intermediate positions, corresponding to the tip of the VH CDR3 loop, were characterized by extreme degeneracy with 2 striking exceptions of landmark residues: in particular, 150 of 152 cases (98.7%) carried a tryptophan (W) residue at VH CDR3 position 10, whereas 135 of 152 cases (88.8%) carried a glycine (G) residue at VH CDR3 position 13 (supplemental Figure 3B).
Subsequently, we scanned M-CLL subsets for additional traces of stereotypy beyond the VH CDR3, in particular for subset-specific patterns of recurrent SHM as reflected in identical amino acid substitutions at the same positions across the rearranged IGHV gene. Restricted amino acid changes were documented in all major and several minor M-CLL subsets, often with degenerate VH CDR3 motifs (supplemental Tables 8-10; supplemental Figure 4B-C), supporting that they likely represent true CLL variants with shared immunogenetic features rather than mere algorithmically derived sequence clusters.
A detailed account of recurrent SHMs in stereotyped subsets is provided in the supplemental Results.
Immunogenetically related stereotyped CLL subsets: introducing the new concept of satellite subsets
Among major stereotyped subsets, 4 pairs of U-CLL subsets with highly similar VH CDR3 motifs were identified. The first concerned subsets #1 and #99, both comprising cases expressing different IGHV genes of phylogenetic clan I (IGHV1/IGHV5/IGHV7 gene subgroups). The VH CDR3 of these subsets had similar composition but differed in length (13 aa in subset #1 vs 14 aa in subset #99). The second pair concerned subsets #8 and #8B, both utilizing the IGHV4-39 gene, whose major difference again involved the VH CDR3 length (19 aa in subset #8 vs 18 aa in subset #8B) (supplemental Figure 5A). For the third pair, subsets #3C2 and #3C3 both used the IGHV1-69 gene: the identified difference involved the offset of the shared VH CDR3 motif by a single position (supplemental Figure 5B). Finally, subsets #7C2 and #7D3, both expressing the IGHV1-69 gene, differed in both VH CDR3 length (23 vs 24 aa, respectively) and motif offset (by a single position) (supplemental Figure 5C).
Next, we searched for immunogenetic relatives of major subsets across the entire stereotyped fraction and identified 156 distinct minor subsets that showed high similarity and, thus, were considered satellites to 21 major subsets. Altogether, satellite subsets (934 sequences) comprised 3% of the total cohort and 7.5% of the stereotyped fraction. A list of all BcR immunoglobulin sequences belonging to satellite subsets is given in supplemental Table 11. Most satellite subsets were small (median number of 3 sequences), albeit populated subsets were also evident (with up to 49 sequences).
Finally, we assessed whether the grouping of each major subset together with its satellite(s) would have a substantial impact on its relative size (Figure 4). Pronounced increases in subset size were seen in major U-CLL subsets #7D3, #169 and #7C2. In M-CLL, subset #148B exhibited the largest number of satellite subsets, which collectively accounted for 61 sequences (supplemental Results; supplemental Figure 6).
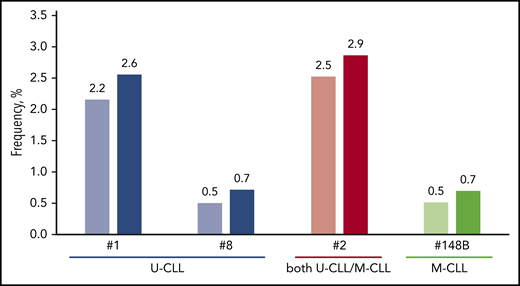
The impact of satellite subsets on the relative size of major stereotyped subsets. More pronounced increases were evident in major subsets comprising U-CLL cases, whereas most M-CLL major subsets were not significantly affected. Characteristic examples concern subsets #1, #2 and #8 that correlate with dismal prognosis. On the other hand, subset #148B showed the highest increase in the category of M-CLL subsets.
The impact of satellite subsets on the relative size of major stereotyped subsets. More pronounced increases were evident in major subsets comprising U-CLL cases, whereas most M-CLL major subsets were not significantly affected. Characteristic examples concern subsets #1, #2 and #8 that correlate with dismal prognosis. On the other hand, subset #148B showed the highest increase in the category of M-CLL subsets.
A final noteworthy case concerned subset #2 that exhibited 8 immunogenetically related subsets all carrying the characteristic landmark residue D at VH CDR3 position 3. One of these subsets was major, namely subset #169 (62 sequences), whereas the others were minor (Figure 5). Grouping together subset #2 (764 sequences) and its relatives (106 sequences) led to a cumulative frequency of 2.9% (870/30 413 sequences).
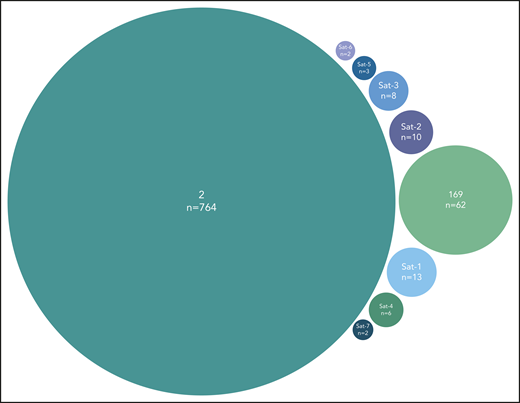
Major subset #2 and its close immunogenetic relatives (satellite subsets). Stereotyped subset #2 had 8 satellite subsets with the most frequent being subset #169, also characterized as major. The remaining satellite subsets were minor and cumulatively accounted for 44 BcR immunoglobulin sequences. Subset #2 satellite full names are the following: #164 (Sat-1), #2B (Sat-2), V3-30|9|1 (Sat-3), V3|11|42 (Sat-4), V3-74|8|1 (Sat-5), V3-48|7|1 (Sat-6), and V3|10|25 (Sat-7).
Major subset #2 and its close immunogenetic relatives (satellite subsets). Stereotyped subset #2 had 8 satellite subsets with the most frequent being subset #169, also characterized as major. The remaining satellite subsets were minor and cumulatively accounted for 44 BcR immunoglobulin sequences. Subset #2 satellite full names are the following: #164 (Sat-1), #2B (Sat-2), V3-30|9|1 (Sat-3), V3|11|42 (Sat-4), V3-74|8|1 (Sat-5), V3-48|7|1 (Sat-6), and V3|10|25 (Sat-7).
To obtain supportive evidence for the relevance of satellite subsets, particularly their clinical significance, if any, we focused on the paradigmatic examples concerning subsets #2 and #169 as well as subsets #1 and #99. In specific, time-to-first-treatment (TTFT) and overall survival analysis for all cases with available data belonging to subsets #2 (212 cases) and #169 (14 cases) showed consistent clinical profiles without any statistical significance (P = .98 and P = .23, respectively). Similar observations were made when considering patients assigned to subsets #1 and #99 for TTFT (139 vs 17 cases, respectively) (P = .45) and OS (153 vs 19 cases, respectively) (P = .65) (Figure 6).

Patients assigned to major stereotyped subsets and their satellites exhibit consistent clinical profiles. Consistent clinical outcomes were observed between patients belonging to subset #2 and subset #169, its main satellite, with no statistical difference regarding: (A) time-to-first-treatment (TTFT) (P = .98) and (B) overall survival (OS) (P = .23). Similarly, patients from stereotyped subset #1 and #99, its main satellite, showed highly similar (C) TTFT (P = .45), and (D) OS (P = .65).
Patients assigned to major stereotyped subsets and their satellites exhibit consistent clinical profiles. Consistent clinical outcomes were observed between patients belonging to subset #2 and subset #169, its main satellite, with no statistical difference regarding: (A) time-to-first-treatment (TTFT) (P = .98) and (B) overall survival (OS) (P = .23). Similarly, patients from stereotyped subset #1 and #99, its main satellite, showed highly similar (C) TTFT (P = .45), and (D) OS (P = .65).
Discussion
BcR immunoglobulin stereotypy has emerged as relevant for unraveling the complex disease biology of CLL, at least for specific patient subgroups,46,47 while also improving patient stratification.34,35,38,41,42,48,49 BcR immunoglobulin stereotypy is not exclusive to CLL, being evident in other mature B-cell malignancies as well.40,50-52 However, BcR immunoglobulin stereotypes are significantly less frequent in these malignancies and display fundamentally different immunogenetic properties compared with CLL. BcR immunoglobulin stereotypy has been also identified in other contexts, like infections53,54 and immune-mediated diseases for example, celiac disease.55 Furthermore, specific B-cell subpopulations have been reported to show an inherently higher tendency to express restricted BcR immunoglobulin.56 These findings underscore the need for further analysis to fully understand the relevance of restricted immune profiles in health and disease.
In this context, we recently scanned the BcR immunoglobulin repertoire of normal B-cell subpopulations in healthy individuals57,58 as well as individuals with monoclonal B-cell lymphocytosis (MBL)59 for the presence of “CLL-specific” stereotypes. We reported that “CLL-specific” stereotyped BcR immunoglobulin represented a small fraction of the repertoire in all B-cell subpopulations studied, thus, questioning the hypothesis for a single normal B-cell progenitor of CLL. Moreover, a very low frequency or even absence of CLL stereotypes related to disease aggressiveness was evident in all settings. These findings highlight the unique immunogenetic signature of CLL, indicating that censoring may normally be exterted on cells expressing “CLL-like” BcR immunoglobulin. Moreover, they emphasize the role of microenvironmental triggering, mediated through the BcR, in CLL ontogeny and evolution.
Our present analysis of BcR immunoglobulin stereotypy in a series of 29 856 CLL patients, 4 times larger than our previous study,15 aimed to refine the immunogenetic characterization of stereotyped subsets as a first, critical step toward identifying potential distinct variants of CLL. Overall, a significant increase in the size of the CLL stereotyped fraction was identified, compared with our previous estimate.15 However, this did not affect the relative frequency of individual subsets, since those defined previously as major retained their status. Arguably, these may represent distinct disease subgroups amenable to compartmentalized research with the potential of overcoming the pronounced heterogeneity of CLL.
To gain better insight into the impact of cohort size on BcR immunoglobulin stereotypy, we performed random data subsampling and built cohorts of different size. In line with previous studies,11-15 the frequency of stereotypy correlated with cohort size, accomplished through the formation of more subsets without any change in the average subset size. Still, the rate by which the frequency of stereotypy increased was not constant but gradually decreased as the analysis progressed to larger cohorts. This finding supports the notion that not all CLL cases are stereotyped, while still consistent with different ontogenetic trajectories for stereotyped and nonstereotyped cases.14,60 Arguably, the stereotyped fraction of CLL might be largely dependent on the functional properties of the clonotypic BcR immunoglobulin, whereas other factors, that is, genomic aberrations, could drive disease pathogenesis in the nonstereotyped group.
Subsequently, we assessed the relationship between BcR immunoglobulin stereotypy and other critical immunogenetic characteristics of the clonotypic BcR immunoglobulin, such as IGHV gene usage and SHM status. We report that the usage of 2 or more different, albeit phylogenetically related, IGHV genes in cases assigned to the same subset is relatively frequent in CLL, recalling findings from other contexts, particularly infections, for example, (1) CD4-mimetic broadly neutralizing antibodies against HIV-1 that use the IGHV1-2 and IGHV1-46 genes61-63 ; (2) influenza-virus receptor binding site-directed antibodies that express the IGHV4-59, IGHV4-39, IGHV4-38-1, and IGHV4-30-2 genes64 ; (3) anti-influenza antibodies against the hemagglutinin stalk that are preferentially encoded by the IGHV1-69 and the highly related IGHV1-18 gene65 ; (4) potent neutralizing antibodies against SARS-CoV-2, which are encoded by the IGHV3-53 and IGHV3-66 genes.66 A possible explanation of this finding, plausible for both CLL but other contexts as well, could be that related IGHV genes have either germline-encoded (U-CLL) or SHM-acquired (M-CLL) specific residues that are crucial for antigen recognition. Alternatively, different IGHV genes may adopt similar conformations in the 3-dimensional (3D) space. This notion is supported by published evidence regarding shape-based clustering of 3D immunoglobulin models, where subset #1 cases utilizing different, yet phylogenetically related, IGHV genes displayed highly similar stuctures.67
The clinical importance of SHM status for clinical decision making in CLL is reflected in the guidelines for disease management provided by the iwCLL in 2018, which mandate that this information is essential before deciding about treatment in all CLL patients both in general practice as well as in clinical trials.43 In our previous study,15 we observed that stereotyped rearrangements were enriched in U-CLL and the same has held in the present analysis. This might reflect a distinct cellular derivation for U-CLL vs M-CLL, whereby the former may correspond to cell subpopulations with inherently restricted BcR immunoglobulin repertoires, capable of recognizing a broad range of antigens due to the expression of certain IGHV genes, provided that these are retained in germline configuration, that is, devoid of SHM.68-70 In that respect, the BcR IGs in U-CLL recall natural autoantibodies that are almost exclusively unmutated and display restricted immunogenetic features.68-71
U-CLL subsets displayed a high level of conservation across the entire VH CDR3, essentially deriving from the utilization of the same IGHV, IGHD, and IGHJ genes. On the other hand, many M-CLL subsets were characterized by degenerate VH CDR3 motifs with few “landmark” (ie, highly conserved) residues, albeit they also displayed remarkably restricted, “subset-biased” SHM patterns. Again, these findings are strongly reminiscent of the immunoglobulin repertoires in viral infections: for instance, in many influenza-virus receptor binding site-directed antibodies, viral engagement appears to depend on the presence of “landmark” residues (ie, just a single dipeptide within the VH CDR3).64
Last, we searched for more distant sequence relatives of major BcR immunoglobulin stereotypes in CLL. The rationale behind this analysis was largely based on accumulating evidence that BcR immunoglobulin encoded by distinct yet phylogenetically related IGHV genes can recognize similar antigenic elements in various contexts, particularly infections, as mentioned above; and, preliminary evidence from our previous study15 strongly alluding to the existence of immunogenetically related subsets: some of these were subsequently found to share similar landscapes of genomic aberrations (eg, subset #2 and its satellite subset #169 have been found to exhibit a strikingly higher frequency of SF3B1 gene mutations compared with the general CLL cohort).72,73 Immunogenetic relations were identified between major subsets as well as between major and minor subsets, mostly concerning U-CLL. Indeed, many major U-CLL subsets had satellite subsets, often large in size, whereas most major M-CLL subsets exhibited few connections, perhaps due to the high level of specificity of their VH CDR3 shaped by SHM. Overall, the grouping of major subsets along with their satellites would lead, at least in certain cases, to a significant increase in their relative size.
Even though the status of satellite subsets could be considered as rather “provisional,” we here provide clinical evidence supporting their relevance, as exemplified by the finding of similar TTFT and OS in stereotyped subset #2 and its satellite subset #169 as well as subset #1 and its satellite subset #99. Admittedly, further research is clearly warranted, however, this preliminary evidence indicates that the identification of satellite subsets could have important implications for biological as well as clinical research in CLL aimed at dissecting disease heterogeneity toward the identification of distinct profiles that would assist in clinical decisions.
BcR stereotypy studies in CLL have focused mainly on the analysis of the VH CDR3, a choice justified by the critical role of this part of the BcR immunoglobulin in antigen recognition, at least in the preimmune repertoire.74 That said, we and others have offered evidence that BcR immunoglobulin light chains are also involved in antigen selection processes in CLL.75-77 This was further supported by recent studies highlighting a unique biological background for CLL cases expressing the IGLV3-21 gene,78 particularly those carrying distinctive imprints of SHM.79 Our present analysis, although confined to the BcR immunoglobulin heavy chain, appears to corroborate this notion, considering that subsets #2 and #169, the prime example of higher-order connections between stereotyped subsets, both utilize the IGLV3-21 gene and carry restricted VL CDR3. Evidently, further work is required, however, the possibility exists for antibody light-chain–restricted antigen recognition in CLL, similar to what has been reported in other contexts as well.80
In summary, we report a significant increase in the frequency of BcR immunoglobulin stereotypy in by far the largest-to-date CLL cohort, while also offering evidence that not all CLL will end up being stereotyped. Importantly, on the basis of our present findings, major subsets emerge as distinct immunogenetic disease variants that can be robustly identified and further studied with the aim of better understanding the natural history of CLL. Finally, we introduce the new concept of satellite subsets, which argues for even greater restriction in the CLL BcR immunoglobulin repertoire than previously thought. This novel immunogenetic classification scheme has the potential to assist in assessing the clinical impact of BcR immunoglobulin stereotypy through the characterization of the immunogenetic relationship between major subsets and their satellites with obvious implications for refined molecular classification of CLL.
For original data, please e-mail the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the Hellenic Foundation for Research and Innovation (HFRI) and the General Secretariat for Research and Technology (GSRT), under grant agreement no. 336 (Project CLLon); the Hellenic Precision Medicine Network in Oncology; the Swedish Cancer Society; the Swedish Research Council; the Knut and Alice Wallenberg Foundation; Karolinska Institutet; Karolinska University Hospital; Radiumhemmets Forskningsfonder, Stockholm; German Research Foundation (DFG) subprojects B1 and B2 of SFB1074; the Bournemouth Leukaemia Fund; the Kuwait Foundation for Advancement of Sciences (KFAS; research grant PR1713MM03); the Polish Scientific Centre (NCN 2018/29/B/NZ5/02706); the Swiss Cancer League (ID 3746, 4395 4660, and 4705); the European Research Council (ERC) Consolidator Grant CLLCLONE (ID 772051); the Swiss National Science Foundation (ID 320030_169670/1 and 310030_192439), Bern, Switzerland; the Leukemia & Lymphoma Society, Translational Research Program (ID 6594-20); the Associazione Italiana per la Ricerca sul Cancro (AIRC) (IG 15426), the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie Grant (agreement no. 794075); the Momentum Grant of the Hungarian Academy of Sciences (LP-95021); the Hungarian National Research, Development and Innovation Office (grant NVKP_16-1-2016-0004); the Leukaemia and Lymphoma Northern Ireland (NI); the AIRC Milan Project (IG 15397), Grant MH-CZ AZV NV19-03-00091, Project NPUII MEYSCZ (CEITEC2020 LQ1601); University Hospital Brno Project (MH-CZ RVO 65269705); AIRC 5 × 1000 (grant 21198); and the Novo Nordisk Foundation (grant NNF16OC0019302).
Authorship
Contribution: A. Agathangelidis designed the research, performed the experiments, analyzed the data and wrote the manuscript; K. Gemenetzi, M.K., P.B., and T.M. analyzed data; V. Giudicelli, M.-P.L., and S.K. were responsible for the curation of the IGHV-IGHD-IGHJ sequences; K.P., Z.D., X.-J.Y., S.J., C.S., L.B.P., R.C.T., L.-A.S., L.S., E.G., M.A., E.T., B.B., C. Baer, D.B., A.N., A.L.d.S., V. Guido, G.M.-H., A.D., C. Brieghel, S.L., M. Meggendorfer, K.B., M. Ritgen, M.F., C.T., A.V., A.P., M.C., L.B., A.S., K.V., M. Roumelioti, H.S.F., S.V., K. Giannopoulos, L.M., T.K.-D., R.S., C. Bödör, F.F., A.K., I.P., D.R., S.A., P.P., P.C., B.E., D.A., L.F., M. Montillo, L.T., N.S., G.G., P.F.d.C., C.N., E.C., A. Anagnostopoulos, C.P., K.F., M.H., D.O., S.S., C.H., D.J., N.C., and S.P. provided data; A.C., A.W.L., C. Belessi, F.D. R.R., and P.G. supervised research; and K.S. designed the study, supervised research, and wrote the manuscript.
Conflict-of-interest disclosure: S.V. has received honoraria from Bayer, AstraZeneca, and Janssen; A.K. has received research funding from AbbVie, Roche/Genentech, Janssen, and AstraZeneca. D.R. has received honoraria from AbbVie, AstraZeneca, Gilead, Janssen, Verastem, and research grants from AbbVie, Gilead, Janssen, and Cellestia. S.A. has received educational grants from Johnson & Johnson, AbbVie, and Roche. K. Giannopoulos has received honoraria from AbbVie, Janssen, and Roche. L.T. has received honoraria from AbbVie, Roche, Janssen, and Shire. A.V. has received honoraria from Janssen and AbbVie. G.G. has received honoraria from AbbVie, Janssen, Sunesis, and AstraZeneca for advisory boards or speaker’s bureau services. C.N. has received research support, consultancy fees, and/or travel grants from AbbVie, Gilead, Janssen, Roche, CSL Behring, Genmab, Sunesis, and Acerta/AstraZeneca outside this work. K.F. has received honoraria from Roche and AbbVie, and Roche travel grants. S.S. has received honoraria and research support from AbbVie, AstraZeneca, Celgene, Gilead, GlaxoSmithKline, Hoffmann La-Roche, Janssen, and Novartis. R.R. has received honoraria from AbbVie, Illumina, Janssen, and Roche. P.G. has received honoraria from AbbVie, Acerta, BeiGene, Gilead, Janssen, Sunesis, and reseach funding from AbbVie, Gilead, Janssen, Novartis, and Sunesis. K.S. has received honoraria and research support from AbbVie, Janssen, AstraZeneca, and Gilead. The remaining authors declare no competing financial interests.
A complete list of the members of the European Research Initiative on CLL (ERIC) appears in supplemental Table 1.
Correspondence: Kostas Stamatopoulos, Institute of Applied Biosciences, Centre for Research and Technology Hellas, 6th km Charilaou–Thermis, 57001 Thermi, Thessaloniki, Greece; e-mail: kostas.stamatopoulos@certh.gr.
