

Key Points
CLL stereotyped subsets 2 and 169 display shared B-cell receptor structural features, supporting shared antigen selection in their ontogeny.
CLL stereotyped subsets 2 and 169 provide a striking example of higher order restrictions of the immunoglobulin gene repertoire in CLL.

Abstract
Chronic lymphocytic leukemia (CLL) major stereotyped subset 2 (IGHV3-21/IGLV3-21, ∼2.5% of all cases of CLL) is an aggressive disease variant, irrespective of the somatic hypermutation (SHM) status of the clonotypic IGHV gene. Minor stereotyped subset 169 (IGHV3-48/IGLV3-21, ∼0.2% of all cases of CLL) is related to subset 2, as it displays a highly similar variable antigen-binding site. We further explored this relationship through next-generation sequencing and crystallographic analysis of the clonotypic B-cell receptor immunoglobulin. Branching evolution of the predominant clonotype through intraclonal diversification in the context of ongoing SHM was evident in both heavy and light chain genes of both subsets. Molecular similarities between the 2 subsets were highlighted by the finding of shared SHMs within both the heavy and light chain genes in all analyzed cases at either the clonal or subclonal level. Particularly noteworthy in this respect was a ubiquitous SHM at the linker region between the variable and the constant domain of the IGLV3-21 light chains, previously reported as critical for immunoglobulin homotypic interactions underlying cell-autonomous signaling capacity. Notably, crystallographic analysis revealed that the IGLV3-21–bearing CLL subset 169 immunoglobulin retains the same geometry and contact residues for the homotypic intermolecular interaction observed in subset 2, including the SHM at the linker region, and, from a molecular standpoint, belong to a common structural mode of autologous recognition. Collectively, our findings document that stereotyped subsets 2 and 169 are very closely related, displaying shared immunoglobulin features that can be explained only in the context of shared functional selection.
Introduction
The existence of stereotyped B-cell receptor immunoglobulin (BcR IG) in chronic lymphocytic leukemia (CLL) strongly implicates antigen selection in disease ontogeny.1-11 Stereotyped cases are classified in multiple subsets of variable size with distinct BcR IG configuration.10 Certain major subsets have emerged as distinct clinical subgroups, exemplified by subset 2, the largest in CLL, accounting for ∼2.5% to 3% of all patients and ∼5.5% of patients requiring treatment.10,12,13
The particular BcR IG of subset 2 is composed of heavy and light chains encoded by the IGHV3-21 and the IGLV3-21 genes, respectively. The clonotypic IGHV3-21 genes bear a variable somatic hypermutation (SHM) load, with most cases classified as mutated (M-CLL), but some cases are unmutated (U-CLL).6,7,10,12,14 Subset 2 BcR IGs display additional distinctive immunogenetic features, including conservation of certain positions in the variable heavy (VH) and variable light (VL) complementarity determining region-3 (CDR3) and recurrent SHMs.15,16 Moreover, they are capable of self-association leading to cell-autonomous signaling that is critically dependent on the substitution of glycine (G) to arginine (R) introduced by SHM at the λ VL-CL linker region in all subset 2 cases analyzed thus far.7,17 From a clinical perspective, it is noteworthy that, independent of SHM status, subset 2 cases have a particularly dismal clinical outcome,12,13,18 similar to that of patients with TP53 aberrations, despite very rarely harboring such aberrations.12,18-24
We have demonstrated that stereotyped subset 169, a minor CLL subset (∼0.2% of all CLL), bears striking immunogenetic similarities to subset 2. More specifically, subset 169 carries: (1) clonotypic heavy chains encoded by the IGHV3-48 gene, which is closely similar to the IGHV3-21 gene; (2) highly similar VH CDR3 motif; (3) clonotypic light chains encoded by the IGLV3-21 gene; (4) both M-CLL and U-CLL cases.10
To obtain comprehensive insight into the ontogenetic relationship and evolution of CLL subsets 2 and 169, we performed next-generation sequencing (NGS) of the respective BcR IG complemented by crystallographic analysis of the BcR Fab fragments. Our findings cement the structural relatedness of the BcR IG of these subsets, providing further evidence for the role of shared antigen selection throughout their natural history.
Materials and methods
Patient group
The patient group comprised 44 patients diagnosed with CLL, according to the International Workshop on CLL/National Cancer Institute guidelines.25 Thirty-one of 44 CLL cases expressed stereotyped BcR IGs assigned to subset 2 (M-CLL, n = 19; U-CLL, n = 12), 7 to subset 169 (M-CLL, n = 4; U-CLL, n = 3), whereas the remaining 6 CLL cases (M-CLL, n = 4; U-CLL, n = 2) were not assigned to stereotyped subsets and were selected for comparisons on the basis of using the IGLV3-21 gene (supplemental Table 1, available on the Blood Web site). The study was approved by the local Ethics Review Committees of the participating institutions and was conducted in accordance with the Declaration of Helsinki.
NGS of IGHV-IGHD-IGHJ and IGLV-IGLJ gene rearrangements
Total cellular RNA was isolated from peripheral blood mononuclear cells. From 1 case assigned to subset 2, we isolated genomic DNA for comparative studies.
Amplification of the IGHV-IGHD-IGHJ and IGLV-IGLJ gene rearrangements was performed by Platinum Taq DNA Polymerase High Fidelity (ThermoFisher Scientific, Waltham, MA) using IG subgroup-specific leader primers as well as IGHJ and IGLC primers for the heavy and λ light chains, respectively, as described.17 PCR products were gel purified (Qiagen, Hilden, Germany), and library preparation was performed according to the manufacturer’s instructions (NEB Next Ultra II DNA Library Prep Kit for Illumina; NEB, Ipswich, MA). Paired-end NGS was performed with the MiSeq Reagent Kit v3 (2 × 300 bp) on the MiSeq Benchtop Sequencer (Illumina).
To assess possible biases related to substrate usage (genomic DNA [gDNA] vs complementary DNA [cDNA]) and different laboratory personnel, we included 2 controls: (1) sample replicates (ie, 1 subset 2 case was analyzed twice starting from either genomic DNA or complementary DNA), and (2) user replicates (ie, 2 libraries from the same subset 2 case prepared by different laboratory members).
Detailed information about the high-throughput sequencing approach is provided in the supplemental Methods.
Bioinformatics analysis and definitions used in this study
Base-calling, quality control, adapter trimming, and demultiplexing were performed with the Illumina signal-processing software. Downstream analysis was performed by the TRIP bioinformatics analytical toolbox.26,27 Only productive, in-frame gene rearrangements were evaluated. SHM characteristics were evaluated as described.7,16,28,29
Clonotypes were defined as IGHV-IGHD-IGHJ or IGLV-IGLJ gene rearrangements with identical IGHV/IGLV gene and VH/VL CDR3 amino acid sequences within a sample. Clonotypes with different amino acid substitutions within the sequence of the rearranged IGHV/IGLV gene excluding the VH/VL CDR3 region were defined as subclones. They were considered to be expanded (shaping clusters) when they included at least 2 sequences, otherwise referred to as a “singleton.” The most expanded clonotype within a sample was deemed the dominant one. Analysis of clonotype branching introduced by SHM was performed using the TRIP tool to determine the 20 most frequent clonotypes of each sample.27
“Convergent recombination” is the term used for describing the phenomenon of different nucleotide sequences degenerately encoding the same VH/VL CDR3 amino acid sequence.30-33
Figure 1 provides a schematic of the definitions used in this manuscript for describing immunogenetic variation at different levels.

A detailed account of the bioinformatics approach is given in supplemental Methods and supplemental Table 2.
Crystallographic studies
The subset 169 case P6540 BcR Fab fragment was expressed in recombinant form in HEK293 cells, as previously described,17 then purified and crystallized. Its crystal structure was determined and refined to 3.4 Å resolution (supplemental Table 3).
Full experimental details are presented in the supplemental Methods, also regarding the comparison of the subset 169 case P6540 fragment vs the subset 2 case P11475 BcR Fab fragment, the latter previously reported by our group.17
Statistical analysis and data visualization tools
Descriptive statistics for qualitative parameters included counts and frequency distributions. For quantitative variables, statistical measures included mean, median, and minimum/maximum values. Analysis of variance was used to evaluate the mean differences in clonotypes, subclones, and convergent recombination between sample categories. Post hoc analysis was performed to assess the multiple pairwise comparisons, using the Bonferroni correction and the homogeneity of possible subgroups identified by Duncan’s test. For the statistics regarding the shared clonotypes between different sample categories, Pearson’s χ2 test with Yates’s continuity correction was used. For all comparisons, a significance level of P = .05 was set.
Data visualization was performed in the R environment and with the open-source data visualization framework RawGraphs (https://rawgraphs.io/). Figures for the crystallographic assays were generated with Pymol (http://www.pymol.org).
Results
Basic information about the NGS run
Overall, 72 PCR amplicons were analyzed, including 39 IGHV-IGHD-IGHJ gene rearrangements (subset 2; n = 32 of which 2 represented duplicates; subset 169, n = 7); and 33 IGLV-IGLJ gene rearrangements (subset 2, n = 21; subset 169, n = 6; nonsubset, n = 6) (supplemental Table 4). Of the aforementioned 39 IGHV-IGHD-IGHJ gene rearrangements, 38 were amplified using cDNA and only 1 by gDNA for the control experiments; detailed information about the results of the control experiments (duplicates) is provided in the supplemental Results.
Overall, 12 925 971 productive gene rearrangement sequences were obtained, corresponding to 4 692 402 IGHV-IGHD-IGHJ (median, 126 681 per sample) and 8 233 569 IGLV-IGLJ gene rearrangement sequences (median, 230 769 per sample) (supplemental Tables 5 and 6).
Clonal composition
We identified 1932 and 3147 unique IGHV-IGHD-IGHJ and IGLV-IGLJ BcR IG clonotypes, respectively. Of these, 1267 (65.6%) heavy chain and 1360 (43.2%) light chain clonotypes were expanded, whereas the remainder were singletons.
In subset 2, each analyzed IGHV-IGHD-IGHJ gene rearrangement carried a median of 81 distinct clonotypes (range, 17-152); the dominant clonotype had a median frequency of 97.7% (range, 66.5%-99.0%). IGLV-IGLJ gene rearrangements carried a higher number of distinct clonotypes per sample compared with their partner heavy chains (median, 216; range, 120-306); the dominant clonotype had a median frequency of 97.2% (range, 73.7%-97.8%) (Figures 2 and 3; supplemental Tables 5 and 6).

Box plots representing the clonality of all sample categories based on the distinct clonotypes identified per sample. Statistical analysis of the clonality patterns revealed that the IGLV-IGLJ gene rearrangements of subset 2 were significantly (P < .05) more diverse than those in all other data sets, whereas those of subset 169 and the nonsubset IGLV3-21 cases did not differ from each other and could form a homogeneous group. The IGHV-IGHD-IGHJ gene rearrangements of subsets 2 and 169 did not differ from each other either, but significantly differed from all LC categories (P < .05). For detailed results of the statistical analysis see supplemental Table 7. HC, heavy chain; LC, light chain.
Box plots representing the clonality of all sample categories based on the distinct clonotypes identified per sample. Statistical analysis of the clonality patterns revealed that the IGLV-IGLJ gene rearrangements of subset 2 were significantly (P < .05) more diverse than those in all other data sets, whereas those of subset 169 and the nonsubset IGLV3-21 cases did not differ from each other and could form a homogeneous group. The IGHV-IGHD-IGHJ gene rearrangements of subsets 2 and 169 did not differ from each other either, but significantly differed from all LC categories (P < .05). For detailed results of the statistical analysis see supplemental Table 7. HC, heavy chain; LC, light chain.
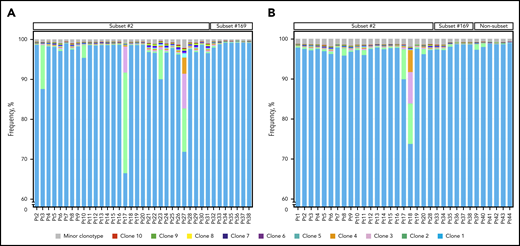
The clonal composition of all heavy chain and IGLV3-21 samples of the cohort is presented. (A) Clustered bar plots depicting the 10 most expanded clonotypes of all heavy chain samples of the present cohort in descending frequency. The relative frequency of each clonotype is computed based on the reads corresponding to the particular clonotype in relation to the total number of reads per sample. The color code represents the ranking of major clonotypes based on their relative frequency. The scale of the graph starts from 60% frequency so that the less expanded clonotypes can be discernible. (B) Clustered bar plots depicting the 10 most expanded clonotypes of all IGLV3-21 samples of the present cohort in descending frequency, as in panel A. Pt, patient.
The clonal composition of all heavy chain and IGLV3-21 samples of the cohort is presented. (A) Clustered bar plots depicting the 10 most expanded clonotypes of all heavy chain samples of the present cohort in descending frequency. The relative frequency of each clonotype is computed based on the reads corresponding to the particular clonotype in relation to the total number of reads per sample. The color code represents the ranking of major clonotypes based on their relative frequency. The scale of the graph starts from 60% frequency so that the less expanded clonotypes can be discernible. (B) Clustered bar plots depicting the 10 most expanded clonotypes of all IGLV3-21 samples of the present cohort in descending frequency, as in panel A. Pt, patient.
In subset 169, each analyzed IGHV-IGHD-IGHJ gene rearrangement carried a median of 54 clonotypes (range, 40-68); the dominant clonotype had a median frequency of 99.1% (range, 97.9%-99.2%). Subset 169 IGLV-IGLJ gene rearrangements carried a higher number of distinct clonotypes per sample compared with their partner heavy chains (median, 144; range, 110-205); the dominant clonotype had a median frequency of 98.3% (range, 97.2%-98.6%; Figures 2 and 3, supplemental Tables 5 and 6).
IGLV3-21–expressing CLL cases that did not belong to either subset 2 or 169 carried a median of 119 distinct clonotypes within their clonotypic IGLV-IGLJ gene rearrangement (range, 100-150); the dominant clonotype had a median frequency of 98.7% (range, 97.2%-98.9%; Figures 2 and 3; supplemental Tables 5 and 6).
Statistical analysis of the clonality patterns revealed that the IGLV-IGLJ gene rearrangements of subset 2 were significantly (P < .05) more diverse from all other data sets, whereas those of subset 169 and the non-subset IGLV3-21 cases did not differ from each other. The clonality patterns of IGHV-IGHD-IGHJ gene rearrangements of subsets 2 and 169 did not differ either, but differed significantly from all light chain categories (P < .05; supplemental Table 7).
Clonotype sharing in subsets 2 and 169
Heavy chains
In subset 2, 415 distinct clonotypes were shared by 2 or more cases (415 of 1618 unique clonotypes; 25.6%). Eight of 415 clonotypes were shared by at least 10 (range 10-19) of the 30 analyzed patients; these were dominant in 1 to 5 patients and of lower frequency in the remaining cases (Figure 4A; supplemental Tables 8A and 9). In subset 169, 55 clonotypes were shared by 2 to 3 of the 7 analyzed cases (55 of 314 unique clonotypes, 17.5%); most shared clonotypes were of low frequency. Two samples carried the same dominant clonotype (98.8%-99.1%), which was also present in 1 more sample, albeit at a low frequency (supplemental Tables 8B and 10). The difference between the 2 subsets regarding the number of shared clonotypes was statistically significant (P = .0172); however, definitive conclusions cannot be drawn, considering that only 7 subset 169 cases were analyzed vs 30 subset 2 cases.

Inter-individual clonotype sharing. (A) Subset 2 heavy chains. All VH CDR3 detected in the subset 2 cohort are depicted by a bubble or dot. The size of the bubbles reflects the relative frequency of each clonotype based on the total number of reads encoding that particular clonotype across all samples. The VH CDR3 of some of the most expanded clonotypes within the entire cohort are stated in the corresponding bubble, and the number of patients sharing that particular clonotype is indicated in the brackets. Most of the expanded clonotypes are shared among different patients (the full list of the entire heavy chain subset 2 clonotypes is given in supplemental Table 8A; for the shared clonotypes, see supplemental Table 9) (B) IGLV3-21 light chains. The entire sequence data set of IGLV3-21 gene rearrangements with 12-aa-long VL CDR3 cohort is depicted. The size of the bubbles reflects the relative frequency of each clonotype based on the total number of reads encoding that particular clonotype across all samples. Some of the expanded clonotypes of the cohort are labeled, and the number of patients sharing that particular clonotype is indicated in the brackets. Most of the expanded clonotypes are shared among different patients (the full list of the entire LC IGLV3-21 with 12-aa-long clonotypes is given in supplemental Table 11A; for the shared clonotypes, see supplemental Table 12).
Inter-individual clonotype sharing. (A) Subset 2 heavy chains. All VH CDR3 detected in the subset 2 cohort are depicted by a bubble or dot. The size of the bubbles reflects the relative frequency of each clonotype based on the total number of reads encoding that particular clonotype across all samples. The VH CDR3 of some of the most expanded clonotypes within the entire cohort are stated in the corresponding bubble, and the number of patients sharing that particular clonotype is indicated in the brackets. Most of the expanded clonotypes are shared among different patients (the full list of the entire heavy chain subset 2 clonotypes is given in supplemental Table 8A; for the shared clonotypes, see supplemental Table 9) (B) IGLV3-21 light chains. The entire sequence data set of IGLV3-21 gene rearrangements with 12-aa-long VL CDR3 cohort is depicted. The size of the bubbles reflects the relative frequency of each clonotype based on the total number of reads encoding that particular clonotype across all samples. Some of the expanded clonotypes of the cohort are labeled, and the number of patients sharing that particular clonotype is indicated in the brackets. Most of the expanded clonotypes are shared among different patients (the full list of the entire LC IGLV3-21 with 12-aa-long clonotypes is given in supplemental Table 11A; for the shared clonotypes, see supplemental Table 12).
A cross-subset comparison revealed no common VH CDR3 clonotypes between the 2 stereotyped subsets, thus supporting the idea that the finding of shared clonotypes between cases within the same subset reflects a true biological phenomenon rather than contamination during the experimental procedure.
Light chains
Overall, 735 clonotypes were shared between all IGLV-IGLJ rearrangements with a 12-aa-long CDR3 (subset 2, n = 21; subset 169, n = 6; nonsubset, n = 3) vs only 9 that were shared between nonsubset IGLV3-21–expressing cases with an 11-aa-long CDR3 (n = 3; P < .001; however, as for the heavy chains, definitive conclusions cannot be drawn, given the great difference in the number of cases analyzed; 30 vs 3). Seventeen of 735 shared clonotypes were highly prevalent, being detected in 20 to 30 samples: 2 of these, QVWDSSSDHPWV and QVWDSGSDHPWV, were not only the most frequently used clonotypes within the entire data set, with relative frequencies of 35.3% and 28.3%, respectively, but also predominated in 12 and 10 samples, respectively (Figure 4B; full lists of the IGLV3-21 clonotypes are given in supplemental Table 11; shared clonotypes are given in supplemental Tables 12 and 13).
Higher intraclonal diversification in the light vs the heavy chain gene rearrangements of subsets 2 and 169
In subset 2, the dominant IGHV-IGHD-IGHJ clonotype displayed a median of 3662 subclones per sample (range, 191-11 041), whereas in subset 169, the dominant IGHV-IGHD-IGHJ clonotype had a median of 1803 subclones per sample (range, 1482-6116; P = .479; supplemental Tables 5 and 7). In both subsets, intraclonal diversity was also observed for the dominant IGLV3-21 clonotype of each case, with medians of 7 015 subclones per sample (range, 1 946-11 866) for subset 2 and 5 101 subclones per sample (range, 4 237-10 381) for subset 169 (P = .99). Nonsubset IGLV3-21 gene rearrangements displayed significantly (P < .05) more restricted intraclonal diversification for the dominant clonotype (median: 4163; range, 3404-4620) compared with subset 2 but did not differ in that respect from subset 169 (P = .99; supplemental Tables 6 and 7).
Comparison of the heavy chain vs the light chain gene rearrangements within subsets 2 and 169 revealed more pronounced intraclonal diversification in the latter (P < .05 for subset 2; P = .065 for subset 169; supplemental Table 7).
Also relevant was the analysis of the IGHV-IGHD-IGHJ and IGLV-IGLJ gene junctions at the nucleotide level, which revealed the presence of convergent recombination30-33 in all sample categories that was more pronounced in subsets 2 and 169 vs nonsubset/IGLV3-21–expressing cases, although the difference did not reach statistical significance (supplemental Figure 1; supplemental Tables 5 and 7).
Truly unmutated sequences
Subset 2 IGHV-IGHD-IGHJ gene rearrangement sequences were found to carry subclones with truly unmutated IGHV genes (ie, displaying 100% germline identity). In detail, 8 of 30 IGHV-IGHD-IGHJ gene rearrangements from subset 2 cases carried such truly unmutated subclones. Only patient 12 carried mostly truly unmutated reads (95 746 of 124 689; 76.8%), a finding in line with a germline identity of 99.65% by Sanger sequencing (U-CLL; supplemental Table 1). The remaining 7 cases carried too few truly unmutated sequences (median, 2; range, 1-60). No truly unmutated IGHV-IGHD-IGHJ gene rearrangement sequences were detected in subset 169.
Turning to the IGLV-IGLJ gene rearrangements, 11 of 21 subset 2 cases had sequences with 100% germline identity. Patient 18 was particularly noteworthy in this respect, considering that, despite displaying a germline identity of 96.86% by Sanger sequencing (supplemental Table 1), he was found to carry 19 946 of 38 036 (52.4%) truly unmutated sequences; the remaining 10 cases carried few truly unmutated sequences (median, 2.5; range, 1-44). Light chain gene rearrangements from subset 169 carried even fewer (range, 1-3) truly unmutated sequences, whereas only 1 nonsubset case, with a germline identity of 99.28% by Sanger sequencing, carried 36 truly unmutated sequences.
Shared recurrent somatic hypermutations in subsets 2 and 169
Heavy chain gene rearrangements
A recurrent 3-nucleotide (AGT) deletion was detected in the VH CDR2 of 28 of 30 examined subset 2 cases, leading to the removal of 1 of 5 consecutive serine residues encoded by the IGHV3-21 germline sequence. This change, previously identified by Sanger sequencing as a recurrent SHM in subset 2 (albeit with an incidence of ∼25%),7,15 was clonal in 10 of 30 cases (median frequency, 99.4%; range, 91.8%-99.9%) with a median of 4019 subclones per sample (range, 208-8088) and subclonal in 18 of 20 of the remainder (median frequency, 0.1%, range, 0.0004%-3.1%) with a median of 36 subclones per sample (range, 1-429). The deletion was completely absent in only 2 of 30 IGHV-IGHD-IGHJ samples (supplemental Table 14A).
Τhe 5-serine stretch is also present in the germline VH CDR2 of the IGHV3-48 gene used in the BcR IG of subset 169. Notably, an identical 3-nucleotide deletion (AGT) in the VH CDR2 was found as a recurrent change in all examined subset 169 cases as well, albeit at subclonal level (median frequency, 1.6%; range, 0.004%-4.4%), with a median of 157 subclones per sample (range, 6-286; supplemental Table 14A).
Light chains
The G-to-R (R110) substitution at the VL-CL linker, previously reported as a ubiquitous SHM in subset 2 (Sanger analysis) and shown to be critical for immunoglobulin self-association leading to cell autonomous signaling in this subset,17 was a clonal event in all subset 2 cases (median frequency, 96.7%; range, 68.9%-98.1%; Figure 5). The same amino acid substitution was detected in all subset 169 cases as a clonal event with a median frequency of 98.6% (range, 96.8%-98.9%). R110 was also present in 4 of 6 nonsubset IGLV3-21 cases as a clonal event (median frequency, 98.5%; range, 96.7%-98.8%); the remaining 2 samples carried the G-to-R substitution at subclonal level (median frequency, 0.01%; range, 0.009%-0.01%; supplemental Table 14B). Τhe presence of the G-to-R substitution was also validated by flow cytometry using an anti-IGLV3-21/R110 antibody and revealing high expression in the 4 cases with clonal G-to-R substitution and low expression in the remaining 2 cases carrying this change at a low-subclonal level.34
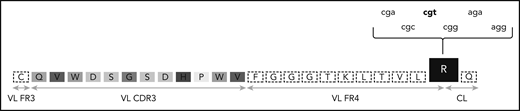
The G-to-R substitution at the VL-CL linker region is being degenerately encoded by different nucleotide sequences. One of the most frequent VL CDR3 of subsets 2 and 169 is depicted. The amino acids of the VL CDR3 region are enclosed in boxes of different shades of gray. The R residue at the VL-CL linker region is in a black box and can be encoded by 6 different nucleotide triplets. The prevailing triplet is set in bold. CL, constant light; FR, framework region.
The G-to-R substitution at the VL-CL linker region is being degenerately encoded by different nucleotide sequences. One of the most frequent VL CDR3 of subsets 2 and 169 is depicted. The amino acids of the VL CDR3 region are enclosed in boxes of different shades of gray. The R residue at the VL-CL linker region is in a black box and can be encoded by 6 different nucleotide triplets. The prevailing triplet is set in bold. CL, constant light; FR, framework region.
Overall similar results were also obtained for the serine (S)-to-G substitution at position 6 within the VL CDR3 which was detected in all examined subset 2 and subset 169 cases at either clonal or subclonal level (supplemental Table 14C).
For both heavy and light chain gene rearrangements, the frequency of mutations at randomly chosen positions (supplemental Tables 14D and 14E) was very different from those reported earlier in this section, further supporting the concept that the findings regarding the recurrent SHMs represent a true biological fact rather than a sequencing artifact.
Highly similar BcR homotypic interactions in subsets 2 and 169
To assess whether the BcR IG from subsets 2 and 169 may share molecular features that parallel their primary structure similarity, we determined the crystal structure of the Fab fragment from the subset 169 P6540 clinical case, which confirmed that subsets 2 and 169 share a similar fold and a conserved modality of intermolecular interaction.
The VH CDR3 loop that defines the CLL subset folded differently in the subset 2 and 169 Fabs, with the latter assuming a more open conformation (Figure 6A-B). These local changes caused a different relative orientation of the VH and VL domains, resulting in a variation of 12° in the domains’ pairing angle (supplemental Table 15). These structural differences affected the shape and chemical characteristics of the antigen-binding site of the 2 BcRs, which, despite maintaining a flat, undulating appearance, displayed a distinct distribution of charged and apolar residues (supplemental Figure 2). The VH CDR3 and pairing orientation between the variable domains apparently did not alter the stability of the heavy-light chain pairing, as judged from the similar buried surface areas (supplemental Table 15).
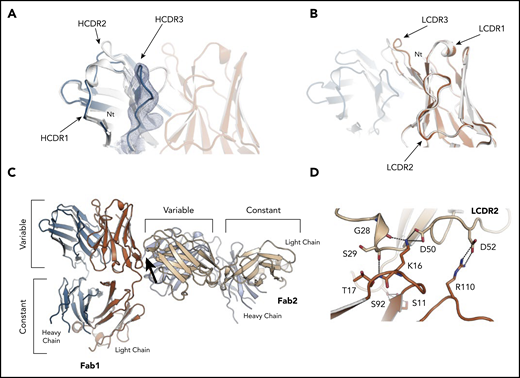
Structure of the subset 169 P6540 case BcR Fab fragment and its homotypic interactions. (A) Superposition of the heavy chain variable domains of P6540 and the subset 2 P11475, showing a different, more open conformation for the VH CDR3 of the P6540 Fab. A cartoon of the P6540 Fab is shown, with heavy chain (blue), light chain (orange), and CDRs labeled accordingly. The subset 2 P11475 Fab is also shown in a cartoon in white. The 2mFo-DFc electron density map is shown from the VH CDR3 loop (blue) and at a contour level of 1.0σ. (B) Superposition of the light chain variable domains of subset 169 P6540 and subset 2 P11475, showing a minor difference in the VL CDR3 conformation caused by a hinged motion of the loop. (C) The homotypic interactions between subset 169 P6540 BCR Fab molecules are revealed by the mutual orientation of 2 interacting Fab molecules in the crystal. Heavy and light chains are shown in the cartoon and are blue and orange, respectively. The interacting region is marked with an arrow. (D) Close-up of the interface between the 2 receptor molecules. The residues involved in the interaction are labeled and shown as sticks. Contacts are dominated by the IGLV3-21 light chain residues through the VL CDR2. All residues shown are also conserved and involved in the homotypic interactions between subset 2 P11475 receptor molecules leading to cell-autonomous signaling.17 Figures were generated with Pymol (http://www.pymol.org).
Structure of the subset 169 P6540 case BcR Fab fragment and its homotypic interactions. (A) Superposition of the heavy chain variable domains of P6540 and the subset 2 P11475, showing a different, more open conformation for the VH CDR3 of the P6540 Fab. A cartoon of the P6540 Fab is shown, with heavy chain (blue), light chain (orange), and CDRs labeled accordingly. The subset 2 P11475 Fab is also shown in a cartoon in white. The 2mFo-DFc electron density map is shown from the VH CDR3 loop (blue) and at a contour level of 1.0σ. (B) Superposition of the light chain variable domains of subset 169 P6540 and subset 2 P11475, showing a minor difference in the VL CDR3 conformation caused by a hinged motion of the loop. (C) The homotypic interactions between subset 169 P6540 BCR Fab molecules are revealed by the mutual orientation of 2 interacting Fab molecules in the crystal. Heavy and light chains are shown in the cartoon and are blue and orange, respectively. The interacting region is marked with an arrow. (D) Close-up of the interface between the 2 receptor molecules. The residues involved in the interaction are labeled and shown as sticks. Contacts are dominated by the IGLV3-21 light chain residues through the VL CDR2. All residues shown are also conserved and involved in the homotypic interactions between subset 2 P11475 receptor molecules leading to cell-autonomous signaling.17 Figures were generated with Pymol (http://www.pymol.org).
Two molecules of the subset 169 receptors interacted homotypically in the crystals through contacts dominated by the IGLV3-21 light chain. The VL CDR2 loop of the combining site was in contact with a conformational epitope that includes the R110 residue (Figure 6C-D). The intermolecular interactions closely resembled what was observed for subset 2 P11475 BcR,17 and confirmed that the IGLV3-21 LC–mediated contacts were retained despite the different, albeit homologous, IGHV gene usage in the 2 receptors and distinct VH CDR3 sequences (supplemental Figure 2). Thus, the IGLV3-21-bearing CLL subset 169 BcR retained the same geometry and contact residues for the homotypic intermolecular interaction observed for subset 217 and, from a molecular standpoint, belonged to a common structural mode of autologous recognition.
Discussion
CLL stereotyped subset 169 is a minor subset bearing BcR IG with striking similarities to CLL stereotyped subset 2,10 alluding to shared antigen selection processes in their pathogenesis. We undertook the present study to explore this hypothesis through a comprehensive analysis of the BcR IG, the critical mediator of antigen interactions in the natural history of CLL clones.
Starting from the immunogenetic profiles determined by NGS, the observed patterns of intraclonal diversification within the immunoglobulin genes were similar in both subsets, revealing a dominant subclone for both the heavy and the light chain clonotypic gene rearrangements. The branching of the clone, reflected in the IG gene amino acid sequences, was relatively extensive in both heavy and light chains, albeit consistently more pronounced in the latter, alluding to their relevance in antigen recognition.16 Further supportive evidence was provided by cross-subset comparisons revealing common clonotypes that were more prevalent for VL CDR3 than for VH CDR3. Admittedly, however, this latter finding may be attributable to the inherently more limited variability of the VL CDR3 compared with the VH CDR3. That cautionary note notwithstanding, our present findings are in line with published evidence regarding the crucial role of the light chains in shaping the structure and, by extension, the function of the BcR IG in CLL.16,35
In subset 2, recurrent SHMs within the IGLV3-21–encoded light chains are linked to the structural properties of the clonotypic BcR IG that allow homotypic BcR IG interactions.17 In more detail, a crucial contribution to the epitope implicated in self-recognition is given by an R residue introduced by SHM at the VL-CL linker position 110. This residue was present in all subset 2 BcR IGs analyzed thus far, and its reversion to the germline-encoded G abrogated cell autonomous signaling.17 Within this context, it is highly relevant that the G-to-R substitution was detected as a clonal event in all subset 2 and 169 IGLV3-21 rearrangements analyzed in this study, strongly indicating shared functional constraint. This conclusion is strongly supported by our crystallographic studies, which revealed homotypic interactions in subset 169 as well, that, similar to those reported for subset 2,17 are dominated by the IGLV3-21 light chain. Of note, the R residue at the VL-CL linker region position 110, a ubiquitous SHM in both subsets, was found to play a critical role in these interactions, in subset 169, as well.
The presence of G-to-R mutation in the nonsubset IGLV3-21 cases analyzed at both clonal and subclonal level further extends previous findings about the seminal role of light chains in antigen selection in CLL, while also raising the intriguing hypothesis of light chain dominance in a substantial fraction of IGLV3-21+ CLL that should be formally proven by future research.16,35,36 In this context, it is also worth mentioning the enrichment for IGLV3-21 expression in a particular epigenetic subgroup of CLL, the “intermediate-programmed CLL epitype,”37,38 highlighting the distinctive nature of CLL cases expressing this particular germline specificity. Further support for this notion is offered by published evidence that IGLV3-21 CLL displays a distinct gene expression profile and a poor clinical course regardless of IGHV gene usage, SHM status and classic cytogenetic abnormalities.39
Our study provides additional evidence for shared antigen selection in the natural history of subsets 2 and 169 through the examination of the SHM patterns in the heavy chain gene rearrangements of the subsets. More particularly, a recurrent 3-nucleotide (AGT) deletion was detected in the VH CDR2 of 28 of 30 subset 2 cases (10 as a clonal and 18 as a subclonal event) and in all subset 169 cases (all subclonal), leading to the removal of 1 of 5 consecutive serine residues encoded by the germline sequence of both the IGHV3-21 and the IGHV3-48 genes. This finding is noteworthy, given the overall low incidence of deletions and insertions introduced by SHM within productive IG gene rearrangements in normal or pathologic B-cell repertoires (with the notable exception of HIV-1 broadly reactive neutralizing antibodies).15,40 We have reported that the VH CDR2 deletion in subset 2 could be accommodated without significantly affecting the local structure,15 although, admittedly, formal proof of the role of this deletion is still pending. That notwithstanding, the very high incidence of this deletion in both subsets, reported here for the first time to our knowledge, argues for shared antigenic drive leaving an identical SHM imprint on the respective heavy chains encoded by 2 phylogenetically related IGHV genes.
An unexpected finding of our study concerns the detection of (mostly) rare subclones lacking any SHM within the IGHV/IGLV genes, even in cases with a considerable SHM load. This rather enigmatic result poses several questions regarding the precise pathogenesis of subsets 2 and 169, alluding to more complex immune trajectories than previously thought, while also prompting research that may reveal whether it is subset specific or a more general phenomenon in CLL.
From a clinical perspective, accumulating evidence suggests that BcR IG stereotypy refines risk stratification in CLL, in certain cases superseding the binary classification into U-CLL and M-CLL.12,13,41 Perhaps the strongest support of this claim is offered by subset 2, characterized by a very aggressive clinical course, even though most patients within this subset are classified as M-CLL.13 Of note, a recent meta-analysis of sequential prospective chemoimmunotherapy trials by the German CLL Study Group concluded that subset 2 membership is an independent adverse-prognostic indicator irrespective of IGHV mutational status and should be proposed for risk stratification of patients.42 On these grounds, the immunogenetic similarities between subsets 2 and 169 argue for their coclustering, with obvious implications for dissecting the heterogeneity of CLL and implementing tailored management approaches. This argument is supported by published evidence of an overall similar background of genomic aberrations in subsets 2 and 169 (eg, both displaying significant enrichment for SF3B1 mutations compared with generic CLL cohorts)20,21 and by our present finding of similar time to first treatment and overall survival (P = .98 and P = .23, respectively) when comparing subsets 2 and 169 (233 vs 14 subset 169 cases with available data from our consortium).
In summary, the immunogenetic and structural profiles reported herein cement the relatedness of subsets 2 and 169, highlighting restricted features at both clonal and subclonal level that can be explained only in the context of shared functional selection. The structural similarities of homotypic interactions identified for subsets 2 and 169 support that the same BcR IG archetype may be shared by cases using different IGHV genes albeit with common ancestry, as in the case of IGHV3-21 and IGHV3-48.
Our high-throughput data sets have been deposited in the public repository, European Nucleotide Archive (ENA) of EMBL-EBI (accession number PRJEB36589). The structure factors and coordinates of the P6540 Fab have been deposited with the Protein Data Bank (accession number 6ZTD).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Theodoros Moysiadis for assisting with the statistical analysis and the staff of the European Synchrotron Radiation Facility (ESRF) and the European Molecular Biology Laboratory (EMBL)-Grenoble for assistance and support in using beamline ID30-A1.
This project was supported by the Hellenic Foundation for Research and Innovation (HFRI) and the General Secretariat for Research and Technology (GSRT), under a grant agreement (project code 82001) for the completion of the studies of K.G. The work was also funded by the ERA-NET on Translational Cancer Research (TRANSCAN-2) Novel project code (MIS) 5041673 (A.C.); the KRIPIS action ODYSSEAS, (MIS) 5002462 (K.S., A.C.); Asklepios Grant Programme funding from Gilead Hellas (K.G., A.C.); the Swedish Cancer Society, the Swedish Research Council; the Knut and Alice Wallenberg Foundation, Karolinska Institutet, Karolinska University Hospital; Radiumhemmets Forskningsfonder, Stockholm (L.-A.S., R.R.); the Ministry of Health, a Czech Republic grant for conceptual development of research organization (FNBr 65269705) (K. Plevova, S.P.); and the Ministry of Education, Youth, and Sports, Czech Republic, project no. CEITEC 2020 (LQ1601) (K. Plevova, S.P.). Support was received from Worldwide Cancer Research grant 19-0096 (M.D.); Bando della Ricerca Finalizzata 2018, Ministero della Salute, Roma, Italy (progetto RF-2018-12368231) (M.D., P.G.); an Investigator Grant 20246 and Special Program on Metastatic Disease-5 per mille 21198 funded by Associazione Italiana per la Ricerca sul Cancro (AIRC, Milano, Italy) (P.G.); and a Marie Sklodowska-Curie individual fellowship (Grant Agreement No. 796491), funded by the European Union’s Horizon 2020 research and innovation program (M.G.).
Authorship
Contribution: K.G. performed the experiments, analyzed the data, and wrote the manuscript; F.P. and M.T. assisted in the bioinformatics analysis and with data visualization; A.A.C., M.G., and C.M. performed the crystallographic experiments; K. Pasentsis assisted with the experiments; K. Plevova, L.-A.S., A.A., R.R., F.D., and S.P. provided materials and patient samples; P.B. provided the data; R.S. assisted in the research and interpretation of results; P.G., K.S., M.D., and A.C. designed and supervised the research and wrote the manuscript; and all authors provided final approval of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Anastasia Chatzidimitriou, Institute of Applied Biosciences, Centre for Research and Technology Hellas, 6th km Charilaou-Thermi Rd, 57001 Thermi, Thessaloniki, Greece; e-mail: achatzidimitriou@certh.gr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal