

Key Points
Canonical WNT signaling is activated in human and murine sclGVHD to promote leukocyte infiltration, fibroblast activation, and fibrosis.
Inhibition of WNT signaling protects against experimental sclGVHD at well-tolerated doses with only minor effects on GVL.

Abstract
Chronic graft-versus-host disease (cGVHD) is a major life-threatening complication of allogeneic hematopoietic stem cell transplantation. The molecular mechanisms underlying cGVHD remain poorly understood, and targeted therapies for clinical use are not well established. Here, we examined the role of the canonical WNT pathway in sclerodermatous cGVHD (sclGVHD). WNT signaling was activated in human sclGVHD with increased nuclear accumulation of the transcription factor β-catenin and a WNT-biased gene expression signature in lesional skin. Treatment with the highly selective tankryase inhibitor G007-LK, the CK1α agonist pyrvinium, or the LRP6 inhibitor salinomycin abrogated the activation of WNT signaling and protected against experimental cGVHD, without a significant impact on graft-versus-leukemia effect (GVL). Treatment with G007-LK, pyrvinium, or salinomycin almost completely prevented the development of clinical and histological features in the B10.D2 (H-2d) → BALB/c (H-2d) and LP/J (H-2b) → C57BL/6 (H-2b) models of sclGVHD. Inhibition of canonical WNT signaling reduced the release of extracellular matrix from fibroblasts and reduced leukocyte influx, suggesting that WNT signaling stimulates fibrotic tissue remodeling by direct effects on fibroblasts and by indirect inflammation-dependent effects in sclGVHD. Our findings may have direct translational potential, because pyrvinium is in clinical use, and tankyrase inhibitors are in clinical trials for other indications.
Introduction
Allogeneic hematopoietic stem cell transplantation (alloSCT) represents the standard care for a wide variety of hematologic, genetic, and immunologic disorders.1 Immune reactions of the transplanted graft against tumor cells primarily contribute as graft-versus-leukemia (GVL) or graft-versus-tumor reactions to the therapeutic effects of alloSCT. However, a major complication of alloSCT is the development of graft-versus-host disease (GVHD).2 GVHD is caused by an impaired tolerance associated with donor T-cell recognition of polymorphic histocompatibility antigens in host tissues, resulting in chronic inflammation and subsequent profound structural remodeling of affected tissues. Although effective therapies are available for the treatment of acute GVHD, therapeutic options for chronic GVHD (cGVHD) remain limited, and cGVHD contributes significantly to the nonrelapse mortality after alloSCT in affected patients.3-6 In particular, sclerodermatous cGVHD (sclGVHD) is associated with impaired quality of life and physical functioning and remains particularly resistant to treatment.
sclGVHD is characterized by profound accumulation of collagen and other components of the extracellular matrix, resulting in progressive tissue fibrosis. The deposition of extracellular matrix in sclGVHD is caused by activation of fibroblasts. In response to profibrotic mediators released from infiltrating leukocytes, resting fibroblasts differentiate into myofibroblasts, which express contractile proteins and release abundant amounts of extracellular matrix. Myofibroblasts are key effector cells in fibrotic tissue remodeling and have been described to accumulate in sclGVHD.7,8 Although intensive research efforts yielded major progress in the understanding of the mechanisms of self-recognition and characterization of the subsequent release of proinflammatory mediators in GVHD,9,10 the molecular mechanisms that bridge inflammation and aberrant fibroblast activation to induce fibrotic tissue remodeling in sclGVHD remain poorly understood. Characterization of those mechanisms may offer potential for novel therapeutic approaches, because targeted inhibition of fibroblast activation has shown benefits in other fibrotic diseases, such as systemic sclerosis and idiopathic pulmonary fibrosis.11-13
WNT signaling is well known for its essential role during embryogenesis and in carcinogenesis, but it also plays an important role in tissue homeostasis in adults.14,15 Canonical WNT signaling involves binding of WNT proteins to various frizzled receptors on the cell membrane and stabilization of β-catenin. Signal transmission by frizzled receptors requires low-density lipoprotein receptor-related protein coreceptors (LRP5/6)16,17 to inhibit the so-called “β-catenin destruction complex,” a multiprotein complex comprising AXIN and casein kinase 1α (CK1α) as central regulatory components.18 Activation of CK1α induces phosphorylation of β-catenin to trigger its degradation.19 Tankyrases regulate the activity of the β-catenin destruction complex by promoting AXIN turnover.20 Inhibition of this complex induces accumulation and nuclear translocation of β-catenin and transcription of WNT target genes.21 Aberrant activation of WNT signaling caused by mutations or by altered expression of central regulatory mediators has been implicated in a variety of diseases.22,23 Accumulating evidence points to a key role for canonical WNT signaling in the pathogenesis of fibrotic diseases.24-33 Of particular interest, several lines of evidence point toward a deregulation of WNT signaling in cGVHD. A modulator of the WNT signaling pathway, Dickkopf-related protein 3 (DKK3), has been identified by mass spectrometry analysis as a biomarker for cGVHD.34 Increased expression of secreted frizzled-related protein 4 was observed in sclGVHD.35 Moreover, ingenuity pathway analysis of the plasma from peripheral blood of patients with GVHD and volunteers without GVHD indicated deregulation of WNT/β-catenin signaling in acute GVHD.36 The WNT agonist R-spondin 1 has been shown to ameliorate GVHD by protecting intestinal stem cells37 and by preventing GVHD-induced dysbiosis.38 Modulation of WNT signaling by lithium has also been studied in a proof-of-concept trial in patients with intestinal GVHD.39
These findings may offer avenues for targeted therapeutic intervention: after long being considered to be “undruggable,” several candidates for pharmaceutical intervention have recently been defined within the canonical WNT pathway, and pharmaceutical inhibition of those targets within the WNT pathway are being evaluated in clinical trials in different malignant diseases.
In the present study, we demonstrate that canonical WNT signaling is activated in human and murine sclGVHD. Using 3 small molecule inhibitors with different modes of action (tankyrase inhibition, activation of CK1α, and destabilization of LRP6) and 2 mouse models, we show that pharmacologic inhibition of canonical WNT signaling reduces inflammation, accumulation of T helper 2 (Th2) cells and M2 macrophages, fibroblast activation, and tissue fibrosis in experimental sclGVHD without reducing GVL. These data provide the first evidence that inhibition of canonical WNT signaling may offer therapeutic potential in sclGVHD.
Materials and methods
Murine models of sclGVHD
The B10.D2 (H-2d) → BALB/c (H-2d) and LP/J (H-2b) → C57BL/6 (H-2b) models are well-established models with minor histocompatibility antigen mismatches. Mice develop moderate to severe dermal fibrosis and moderate pulmonary fibrosis.40-42
B10.D2 (H-2d) → BALB/c (H-2d) model
The model was performed as described before.7,8 Briefly, recipient BALB/c (H-2d) mice underwent total body irradiation with 700 cGy. Sixteen hours after irradiation, each recipient BALB/c (H-2d) mouse received 4 × 106 splenocytes and 1.5 × 106 bone marrow cells from BALB/c (H-2d) mice (syngeneic controls) or B10.D2 (H-2d) mice (allogeneic transplantation) by tail vein injection.40,41
LP/J (H-2b) → C57BL/6 (H-2b) model
C57BL/6 (H-2b) recipient mice received 3 × 107 splenocytes isolated from C57BL/6 (H-2b) mice (syngeneic controls) or LP/J (H-2b) mice (allogeneic transplantation) by tail vein injection.43
Treatment with G007-LK, pyrvinium, or salinomycin
Mice were treated with oral G007-LK (20 mg/kg per day), intraperitoneal pyrvinium (10 mg/kg per day), or intraperitoneal salinomycin (5 mg/kg per day). The treatment was started after stable engraftment 10 days post–bone marrow transplantation (BMT) in the B10.D2 (H-2d) → BALB/c (H-2d) or LP/J (H-2b) → C57BL/6 (H-2b) model until mice were euthanized.44
RNA sequencing library construction and bioinformatics analysis
Total RNA was extracted from skin biopsies of patients with cGVHD and from age- and sex- matched healthy volunteers, as described.45 RNA sequencing (RNASeq) was performed by Novogene Co, Ltd (Cambridge, United Kingdom). Thresholds for the identification of differentially expressed genes (DEGs) were false discovery rate–corrected P value <.2 and fold change >1.5. Principal component analysis plots, heat maps, volcano plots, and bubble plots were generated with ggplot2 R packages. DEGs were subsequently used for gene ontology (GO) and gene functional analysis with the clusterProfiler package.
Immunofluorescence
To visualize canonical WNT signaling in fibroblasts, skin sections of sclGVHD and controls were incubated with polyclonal rabbit anti–β-catenin and polyclonal mouse anti–prolyl-4-hydroxylase β (P4Hβ) antibodies for human sections or polyclonal mouse anti-vimentin antibodies for murine sections. The expression of β-catenin and P4Hβ or vimentin was visualized with secondary antibodies labeled with Alexa Fluor 488 or Alexa Fluor 594.
Leukocytes, T cells, and B cells were identified by staining with 4′,6-diamidino-2-phenylindole (DAPI) combined with anti-CD45 antibodies, anti-CD3 antibodies, or anti-B220 antibodies, respectively. Th1 cells were counted as DAPI, CD3, T-bet–positive cells. Th2 cells were counted as DAPI, CD3, GATA3–positive cells.
Macrophages were identified as F4/80 and DAPI double-positive cells. M1 macrophages were counted as DAPI, iNOS, CD11C, and F4/80–positive cells. M2 macrophages were quantified as DAPI, arginase, c-MAF, and F4/80-positive cells. All sections were counted by 2 experienced reviewers in a blinded manner.
Voronoi tessellation of ex vivo immunofluorescence pictures and cell counting was performed using ImageJ2 software and CellProfiler.46,47
Additional detailed information can be found in supplemental Materials and methods (available on the Blood Web site). Patient information is provided in Table 1.
Results
Canonical WNT signaling is activated in sclGVHD
To analyze whether canonical WNT signaling is activated in human sclGVHD, we performed 2 sets of experiments using lesional skin biopsies of patients with sclGVHD after alloSCT and age- and sex-matched healthy individuals (Figure 1A).
![WNT signaling is active in human sclGVHD. (A) Skin sections from healthy volunteers, patients after allogeneic transplantation without GVHD, and patients with cGVHD (original magnification ×100; hematoxylin and eosin stain; scale bars, 250 μm). (B-D) Immunofluorescence analyses of nuclear β-catenin: β-catenin was detected by immunofluorescence staining in skin biopsies of patients with cGVHD, patients with allogeneic transplantation without GVHD, and healthy volunteers. Fibroblasts were identified by staining with P4Hβ. Representative images of patients with cGVHD and controls (original magnification ×400). (B) Nuclear β-catenin expression was increased in the dermis of patients with cGVHD (sclGVHD; n = 4) compared with patients with allogeneic transplantation without GVHD (n = 6) and healthy volunteers (n = 8). Boxes denote the area of panel C. (C) Voronoi tessellation of nuclear β-catenin+ fibroblasts. (D) Percentage of nuclei β-catenin+ fibroblasts in the skin of patients with sclGVHD, patients with allogeneic transplantation without GVHD, and healthy volunteers. (E-G) RNASeq analysis of 6 sclGVHD patients and 4 healthy controls. **P < .05; ***P < .01. (E) Heat map illustration of DEGs. (F) Volcano plot. The expression of each gene is plotted as the log2 fold change in expression against −log10 (false discovery rate [FDR]); the black dotted lines indicate thresholds of FDR, 0.2 and fold change, 1.5. Differentially regulated genes with FDR <0.2 and fold change >1.5 compared with controls are shown in red (upregulated) and green (downregulated). (G) Bubble plots displaying significant enrichment of GO biological processes. The color of the bubble represents the P value, and the size of the bubble represents the number of DEGs in the data sets associated with the GO processes.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/17/10.1182_blood.2020008720/1/m_bloodbld2020008720f1.png?Expires=1765941736&Signature=GWx6WqUwvrBCK8R7Eh8HV24ZDsYa2H6LvmhMABxJ81pf5oDqLMZ25N3ogtMrWV-ruok75JFGLNIJodIExQmsYj0PTFwx0iOij7RF3yr9lDEKJZnR7pdRgOV2Y0rw9is4g7HuwAGAAk-0qA~IZfzmZvl5R9ZFwmhAcFtyNdIb55JWQRdpw0bMQmp0wftlEmDmzp3H6BAsYiXiLPiyEcjSn3Zq-PWOaYQOoMGtaBG5OfvR0uaPGrpI-M1XN3FJJ5Nx5lWQSXe0ZeJ5rVN65GczVifA9TelN2JslGNUmBiKP-DrnvdgyJPs63he2Qt3lvMkk8ICw-4pB-7uZrLKGey90g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
WNT signaling is active in human sclGVHD. (A) Skin sections from healthy volunteers, patients after allogeneic transplantation without GVHD, and patients with cGVHD (original magnification ×100; hematoxylin and eosin stain; scale bars, 250 μm). (B-D) Immunofluorescence analyses of nuclear β-catenin: β-catenin was detected by immunofluorescence staining in skin biopsies of patients with cGVHD, patients with allogeneic transplantation without GVHD, and healthy volunteers. Fibroblasts were identified by staining with P4Hβ. Representative images of patients with cGVHD and controls (original magnification ×400). (B) Nuclear β-catenin expression was increased in the dermis of patients with cGVHD (sclGVHD; n = 4) compared with patients with allogeneic transplantation without GVHD (n = 6) and healthy volunteers (n = 8). Boxes denote the area of panel C. (C) Voronoi tessellation of nuclear β-catenin+ fibroblasts. (D) Percentage of nuclei β-catenin+ fibroblasts in the skin of patients with sclGVHD, patients with allogeneic transplantation without GVHD, and healthy volunteers. (E-G) RNASeq analysis of 6 sclGVHD patients and 4 healthy controls. **P < .05; ***P < .01. (E) Heat map illustration of DEGs. (F) Volcano plot. The expression of each gene is plotted as the log2 fold change in expression against −log10 (false discovery rate [FDR]); the black dotted lines indicate thresholds of FDR, 0.2 and fold change, 1.5. Differentially regulated genes with FDR <0.2 and fold change >1.5 compared with controls are shown in red (upregulated) and green (downregulated). (G) Bubble plots displaying significant enrichment of GO biological processes. The color of the bubble represents the P value, and the size of the bubble represents the number of DEGs in the data sets associated with the GO processes.
WNT signaling is active in human sclGVHD. (A) Skin sections from healthy volunteers, patients after allogeneic transplantation without GVHD, and patients with cGVHD (original magnification ×100; hematoxylin and eosin stain; scale bars, 250 μm). (B-D) Immunofluorescence analyses of nuclear β-catenin: β-catenin was detected by immunofluorescence staining in skin biopsies of patients with cGVHD, patients with allogeneic transplantation without GVHD, and healthy volunteers. Fibroblasts were identified by staining with P4Hβ. Representative images of patients with cGVHD and controls (original magnification ×400). (B) Nuclear β-catenin expression was increased in the dermis of patients with cGVHD (sclGVHD; n = 4) compared with patients with allogeneic transplantation without GVHD (n = 6) and healthy volunteers (n = 8). Boxes denote the area of panel C. (C) Voronoi tessellation of nuclear β-catenin+ fibroblasts. (D) Percentage of nuclei β-catenin+ fibroblasts in the skin of patients with sclGVHD, patients with allogeneic transplantation without GVHD, and healthy volunteers. (E-G) RNASeq analysis of 6 sclGVHD patients and 4 healthy controls. **P < .05; ***P < .01. (E) Heat map illustration of DEGs. (F) Volcano plot. The expression of each gene is plotted as the log2 fold change in expression against −log10 (false discovery rate [FDR]); the black dotted lines indicate thresholds of FDR, 0.2 and fold change, 1.5. Differentially regulated genes with FDR <0.2 and fold change >1.5 compared with controls are shown in red (upregulated) and green (downregulated). (G) Bubble plots displaying significant enrichment of GO biological processes. The color of the bubble represents the P value, and the size of the bubble represents the number of DEGs in the data sets associated with the GO processes.
We first performed immunofluorescence staining for β-catenin, the central integrator of canonical WNT signaling, in skin sections obtained from patients with sclGVHD and age- and sex-matched healthy individuals. Double staining of β-catenin with the fibroblast marker P4Hβ and DAPI demonstrated prominent nuclear staining for β-catenin in >47% of fibroblasts in patients with sclGVHD compared with only 12% in healthy volunteers. Fibroblasts in the skin of patients with allogeneic transplantation, but without GVHD, also did not show nuclear accumulation of β-catenin (Figure 1B-D). Intense nuclear staining was observed in papillary and reticular dermal fibroblasts of patients with sclGVHD. In contrast, only few papillary fibroblasts in the skin of healthy individuals or patients after allogeneic transplantation without GVHD stained positive for nuclear β-catenin. Quantification by Voronoi tessellation confirmed the increased nuclear accumulation of β-catenin in sclGVHD (Figure 1B-D).
To further validate the aberrant activation of canonical WNT signaling in sclGVHD, we next performed RNASeq on additional skin biopsies from patients with sclGVHD and healthy individuals. We found 667 genes to be upregulated and 511 genes to be downregulated by ≥1.5-fold in sclGVHD compared with healthy controls (Figure 1E-F). Despite heterogeneity within the sclGVHD group (supplemental Figure 1A for general gene expression with principal component analysis; supplemental Figure 1A for lead cytokines), GO enrichment analysis of DEGs highlighted characteristic changes in patients with sclGVHD that clearly separated them from controls. These included 3 functional categories: (1) as expected, we observed several biological processes in patients with sclGVHD related to T-cell, B-cell, and macrophage activation, including several processes related to type 2 immune responses (Figure 1G); (2) we detected several biological processes related to fibroblast activation and fibrotic tissue remodeling, including extracellular structure and extracellular matrix organization, epithelial-to-mesenchymal transition, tissue regeneration, and extracellular matrix assembly; and (3) of particular interest, we observed that biological processes related to canonical WNT signaling are upregulated in sclGVHD, including cell-cell signaling by WNT, canonical WNT signaling pathway, and regulation of canonical WNT signaling pathway. Thus, the unbiased RNASeq analysis highlights an activation of canonical WNT signaling as a shared feature among patients with cutaneous sclGVHD.
To determine whether WNT signaling was operational in experimental sclGVHD, we analyzed 2 mouse models using 2 established readouts: nuclear accumulation of β-catenin, the central integrator of canonical WNT signaling, and messenger RNA (mRNA) levels of the prototypical WNT target gene Axin2. β-catenin accumulated in the nuclei of fibroblasts in allografted BALB/c (H-2d) mice compared with syngeneic controls. The number of fibroblasts positive for nuclear β-catenin and the accumulation pattern were comparable to human sclGVHD. The mRNA levels of Axin2 were also upregulated in BALB/c (H-2d) and C57BL/6 (H-2b) mouse models of sclGVHD, demonstrating that these models are suitable to study the role of canonical WNT signaling in sclGVHD.
G007-LK, pyrvinium, and salinomycin inhibit canonical WNT signaling in cultured fibroblasts and in murine sclGVHD
To determine whether the tankyrase inhibitor G007-LK, the CK1α activator pyrvinium, and the LRP6 destabilizing agent salinomycin can effectively inhibit canonical WNT signaling, we first incubated human dermal fibroblasts with G007-LK, pyrvinium, and salinomycin in concentrations of 5 µM, 30 nM, and 1.25 µM, respectively. G007-LK, pyrvinium, and salinomycin effectively inhibited WNT1-induced nuclear accumulation of β-catenin in cultured fibroblasts (supplemental Figure 2A-B). G007-LK, pyrvinium, and salinomycin also reduced the levels of AXIN2 mRNA in dermal fibroblasts compared with healthy dermal fibroblasts (supplemental Figure 2C).
We next evaluated whether treatment with G007-LK, pyrvinium, or salinomycin can also prevent the induction of canonical WNT signaling in experimental sclGVHD. We observed that all 3 WNT inhibitors induced a profound reduction in Axin2 mRNA in the B10.D2 (H-2d) → BALB/c (H-2d) and LP/J (H-2b) → C57BL/6 (H-2b) models of sclGVHD, as well as decreased the numbers of fibroblasts with nuclear staining of β-catenin in the B10.D2 (H-2d) → BALB/c (H-2d) model (supplemental Figures 3 and 4).
The doses of G007-LK, pyrvinium, and salinomycin, which are required for effective inhibition of canonical WNT signaling, were well tolerated. Clinical monitoring did not demonstrate any changes in activity, behavior, texture of the fur, or consistency of the stool compared with vehicle-treated mice. Together, these data demonstrate effective inhibition of canonical WNT signaling by G007-LK, pyrvinium, and salinomycin in well-tolerated doses.
Inhibition of canonical WNT signaling ameliorates clinical signs of experimental sclGVHD
We next aimed to evaluate whether treatment with G007-LK, pyrvinium, or salinomycin can ameliorate clinical features of sclGVHD in the B10.D2 (H-2d) → BALB/c (H-2d) and LP/J (H-2b) → C57BL/6 (H-2b) models. All mice experienced an initial radiation-induced weight loss that peaked within 4 days after allogeneic BMT (alloBMT). However, although syngeneic controls recovered rapidly and constantly increased their body weight throughout the observation period, vehicle-treated allografted mice in both models recovered more slowly and did not show further increases in body weight after 12 days post-alloBMT. Treatment of allografted mice with G007-LK, pyrvinium, or salinomycin partially reduced sclGVHD-induced weight loss. These differences (ie, higher body weights in treated mice compared with mice with allogeneic transplantation) were maintained until the end of the experiment (Figure 2A).
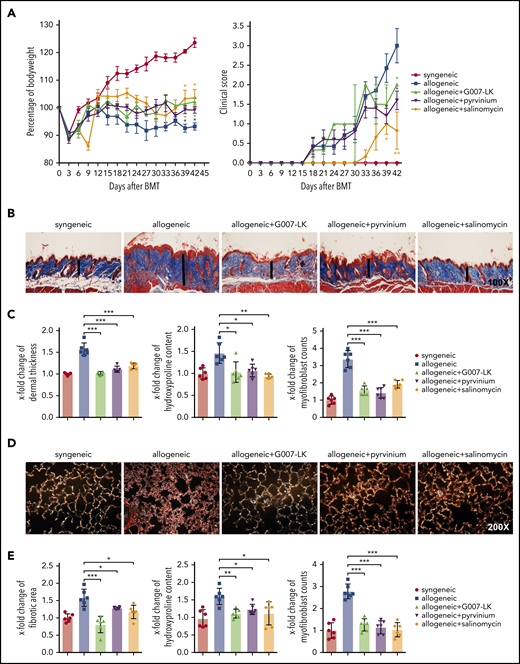
Inhibition of WNT signaling by G007-LK, pyrvinium, or salinomycin prevents the clinical features of sclGVHD in the model of B10.D2 (H-2d) → BALB/c (H-2d). (A) Preventive treatment with G007-LK, pyrvinium, or salinomycin impeded weight loss after alloBMT and resulted in a significantly higher body weight (left panel). The clinical composite score for cutaneous cGVHD was lower in mice treated with G007-LK, pyrvinium, or salinomycin compared with sham-treated mice (right panel). (B) Representative trichrome stained images of murine sclGVHD skin (original magnification ×100). WNT inhibitors prevented the histological changes in the dermis in experimental sclGVHD. Dermal thickness is represented by vertical black lines. (C) WNT inhibitors reduced dermal thickening, decreased the hydroxyproline content in the murine skin, and diminished the differentiation of resting dermal fibroblasts into myofibroblasts compared with vehicle-treated mice with allogeneic transplantation. (D) Representative lung sections (original magnification ×200; Sirius Red stain). (E) Fibrotic area, hydroxyproline content, and myofibroblast counts of murine lung (n = 6 mice per group). *P < .5; **P < .05; ***P < .01.
Inhibition of WNT signaling by G007-LK, pyrvinium, or salinomycin prevents the clinical features of sclGVHD in the model of B10.D2 (H-2d) → BALB/c (H-2d). (A) Preventive treatment with G007-LK, pyrvinium, or salinomycin impeded weight loss after alloBMT and resulted in a significantly higher body weight (left panel). The clinical composite score for cutaneous cGVHD was lower in mice treated with G007-LK, pyrvinium, or salinomycin compared with sham-treated mice (right panel). (B) Representative trichrome stained images of murine sclGVHD skin (original magnification ×100). WNT inhibitors prevented the histological changes in the dermis in experimental sclGVHD. Dermal thickness is represented by vertical black lines. (C) WNT inhibitors reduced dermal thickening, decreased the hydroxyproline content in the murine skin, and diminished the differentiation of resting dermal fibroblasts into myofibroblasts compared with vehicle-treated mice with allogeneic transplantation. (D) Representative lung sections (original magnification ×200; Sirius Red stain). (E) Fibrotic area, hydroxyproline content, and myofibroblast counts of murine lung (n = 6 mice per group). *P < .5; **P < .05; ***P < .01.
sclGVHD occurred in all BALB/c recipients transplanted with B10.D2 bone marrow/splenocytes [B10.D2 (H-2d) → BALB/c (H-2d) mice] and in all C57BL/6 mice receiving splenocytes from LP/J mice [LP/J (H-2b) → C57BL/6 (H-2b) mice], whereas syngeneic controls in both models [BALB/c (H-2d) → BALB/c (H-2d) and C57BL/6 (H-2b) → C57BL/6 (H-2b)] did not show any signs of cGVHD. In addition to weight loss, clinical signs of cGVHD in allografted mice included reduced mobility and diarrhea (Figures 2A and 3A).
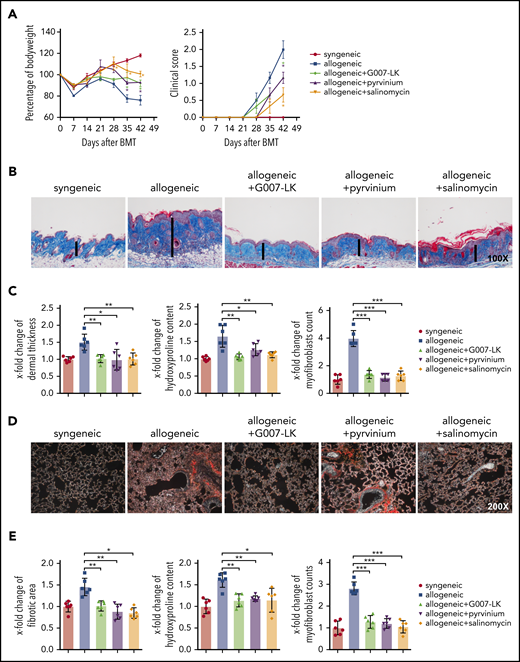
Inhibition of WNT signaling by G007-LK, pyrvinium, or salinomycin prevents the clinical features of sclGVHD in the LP/J (H-2b) → C57BL/6 (H-2b) model. (A) Preventive treatment with G007-LK, pyrvinium, or salinomycin reduced weight loss after alloBMT and resulted in higher body weight (left panel). The clinical composite score for cutaneous cGVHD was lower in mice treated with G007-LK, pyrvinium, or salinomycin compared with vehicle-treated mice (right panel). (B) Representative trichrome-stained images of the skin of sclGVHD mice (original magnification ×100). WNT inhibitors prevented the histological changes in the dermis in experimental sclGVHD. (C) WNT inhibitors reduced dermal thickening, decreased the hydroxyproline content of the skin, and diminished the differentiation of resting fibroblasts into myofibroblasts compared with vehicle-treated mice with allogeneic transplantation. (D) Representative lung sections (original magnification ×200; Sirius Red stain). (E) Fibrotic area, hydroxyproline content, and myofibroblast counts of murine lung (n = 6 mice per group). *P < .5; **P < .05; ***P < .01.
Inhibition of WNT signaling by G007-LK, pyrvinium, or salinomycin prevents the clinical features of sclGVHD in the LP/J (H-2b) → C57BL/6 (H-2b) model. (A) Preventive treatment with G007-LK, pyrvinium, or salinomycin reduced weight loss after alloBMT and resulted in higher body weight (left panel). The clinical composite score for cutaneous cGVHD was lower in mice treated with G007-LK, pyrvinium, or salinomycin compared with vehicle-treated mice (right panel). (B) Representative trichrome-stained images of the skin of sclGVHD mice (original magnification ×100). WNT inhibitors prevented the histological changes in the dermis in experimental sclGVHD. (C) WNT inhibitors reduced dermal thickening, decreased the hydroxyproline content of the skin, and diminished the differentiation of resting fibroblasts into myofibroblasts compared with vehicle-treated mice with allogeneic transplantation. (D) Representative lung sections (original magnification ×200; Sirius Red stain). (E) Fibrotic area, hydroxyproline content, and myofibroblast counts of murine lung (n = 6 mice per group). *P < .5; **P < .05; ***P < .01.
In the B10.D2 (H-2d) → BALB/c (H-2d) model, clinical signs of cutaneous sclGVHD, with skin thickening, xerosis, hyperkeratosis, alopecia, and skin ulcers, became evident 18 days after alloBMT and progressively worsened in vehicle-treated B10.D2 (H-2d) → BALB/c (H-2d) mice. Treatment with all 3 WNT inhibitors ameliorated cutaneous cGVHD, with a reduction in the clinical composite score of cutaneous cGVHD from 3.0 to 0.8, 2.0, or 1.6 in mice treated with G007-LK, pyrvinium, or salinomycin, respectively. Cutaneous cGVHD developed slightly later and was less severe in the LP/J (H-2b) → C57BL/6 (H-2b) model. Treatment with G007-LK, pyrvinium, or salinomycin also improved cutaneous cGVHD in this model, with significant reductions in the composite score of cutaneous cGVHD in G007-LK–, pyrvinium-, or salinomycin-treated LP/J (H-2b) → C57BL/6 (H-2b) mice compared with vehicle-treated allografted LP/J (H-2b) → C57BL/6 (H-2b) mice.
WNT inhibitors reduce fibrotic tissue remodeling in murine sclGVHD
To evaluate the effect of G007-LK, pyrvinium, or salinomycin on histological changes associated with sclGVHD, we next analyzed skin, lung, and intestinal samples of B10.D2 (H-2d) → BALB/c (H-2d) mice and LP/J (H-2b) → C57BL/6 (H-2b) mice. B10.D2 (H-2d) → BALB/c (H-2d) mice developed a systemic sclerosing disease with dermal fibrosis, infiltration of leukocytes, and atrophy of the subcutis. Fibrotic changes were not observed in syngeneic control mice [BALB/c (H-2d) → BALB/c (H-2d)]. The dermal thickness and the hydroxyproline content in B10.D2 (H-2d) → BALB/c (H-2d) mice increased by 1.6 ± 0.17–fold and 1.4 ± 0.3–fold, respectively, after 42 days compared with BALB/c (H-2d) → BALB/c (H-2d) control mice. The number of myofibroblasts was also significantly higher in mice undergoing alloBMT than in syngeneic controls. Treatment with G007-LK, pyrvinium, or salinomycin completely inhibited sclGVHD-induced fibrosis, with dermal thickness, hydroxyproline content, and myofibroblast counts comparable to those in syngeneic BALB/c (H-2d) → BALB/c (H-2d) controls (Figure 2B-C). In addition to skin fibrosis, B10.D2 (H-2d) → BALB/c (H-2d) mice developed pulmonary fibrosis with increased collagen-covered area, hydroxyproline content, and myofibroblast counts in allogeneic mice. WNT inhibition by G007-LK, pyrvinium, or salinomycin also effectively ameliorated cGVHD-induced pulmonary fibrosis (Figure 2D-E).
Dermal and pulmonary fibrosis also developed in LP/J (H-2b) → C57BL/6 (H-2b) mice with pronounced fibrotic remodeling and collagen accumulation compared with C57BL/6 (H-2b) → C57BL/6 (H-2b) control mice. All 3 inhibitors demonstrated potent antifibrotic effects with normalization of dermal thickness, hydroxyproline content, and myofibroblast counts in skin (Figure 3B-C) and of collagen-covered area in lungs, respectively (Figure 3D-E).
Treatment with G007-LK, pyrvinium, or salinomycin also ameliorated the intestinal manifestations of experimental sclGVHD in both mouse models without affecting the numbers of LGR5+ stem cells (supplemental Figure 5).
The inhibitory effects of G007-LK, pyrvinium, or salinomycin on dermal and pulmonary fibrosis and intestinal inflammation were comparable in both models, and all 3 drugs were well tolerated, highlighting the therapeutic potential of WNT inhibition in sclGVHD.
G007-LK, pyrvinium, or salinomycin inhibits fibroblast-to-myofibroblast differentiation in human dermal fibroblasts
We next aimed to assess the direct effects of G007-LK, pyrvinium, or salinomycin on cultured fibroblasts. Cultured human dermal fibroblasts expressed increased levels of myofibroblast marker αSMA and upregulated stress fibers formation compared with cultured normal human dermal fibroblasts (Figure 4A). Consistently, incubation with WNT1 also promoted deposition of extracellular matrix components, such as collagen type I and type III (Figure 4B). Inhibition of WNT signaling by G007-LK, pyrvinium, or salinomycin at concentrations of 5 µM, 30 nM, and 1.25 µM reduced the expression of αSMA, decreased the formation of stress fibers in dermal fibroblasts (Figure 4A), and effectively inhibited fibroblast-to-myofibroblast transition induced by WNT1, with reduced deposition of extracellular matrix (Figure 4B). These data provide evidence for a direct inhibitory effect of WNT inhibition on fibroblast activation.
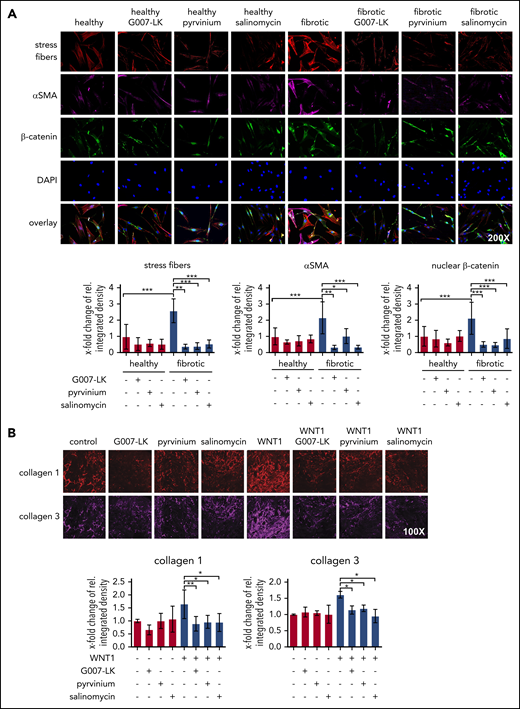
The WNT pathway regulates fibroblast activation. (A) Representative images of immunofluorescence staining for stress fibers, αSMA, and β-catenin in human dermal fibroblasts, with and without WNT inhibitors (original magnification ×200). Quantifications indicate that treatment with WNT inhibitors (24-hour incubation) prevents the upregulation of stress fiber formation, αSMA expression, and β-catenin accumulation in fibroblasts. (B) Representative pictures and quantifications of immunofluorescence staining for collagen type I and III in decellularized extracellular matrixes in human fibroblasts with or without WNT1 and with or without WNT inhibitors (48-hour incubation) (n = 4 cell lines each) (original magnification ×100). *P < .5; **P < .05; ***P < .01.
The WNT pathway regulates fibroblast activation. (A) Representative images of immunofluorescence staining for stress fibers, αSMA, and β-catenin in human dermal fibroblasts, with and without WNT inhibitors (original magnification ×200). Quantifications indicate that treatment with WNT inhibitors (24-hour incubation) prevents the upregulation of stress fiber formation, αSMA expression, and β-catenin accumulation in fibroblasts. (B) Representative pictures and quantifications of immunofluorescence staining for collagen type I and III in decellularized extracellular matrixes in human fibroblasts with or without WNT1 and with or without WNT inhibitors (48-hour incubation) (n = 4 cell lines each) (original magnification ×100). *P < .5; **P < .05; ***P < .01.
G007-LK, pyrvinium, or salinomycin modulates leukocyte infiltration
Inflammatory infiltrates are characteristic features of sclGVHD. The infiltrating leukocytes modulate the activity of resident fibroblasts by the release of profibrotic cytokines. Thus, we investigated whether potential anti-inflammatory effects may contribute to the antifibrotic effects of WNT inhibition in sclGVHD.
Quantification of the number of CD45+ cells demonstrated that treatment with G007-LK, pyrvinium, or salinomycin reduced total leukocyte counts in the skin of B10.D2 (H-2d) → BALB/c (H-2d) and LP/J (H-2b) → C57BL/6 (H-2b) mice. The numbers of T cells, B cells, and macrophages were decreased with all 3 WNT inhibitors (Figure 5A,C; supplemental Figure 6A,C). Further analyses of different T-cell subpopulations demonstrated significant decreases in Th1 and Th2 cells in the skin, with reduced numbers of M1 and M2 macrophage counts upon WNT inhibition (Figure 5B-C; supplemental Figure 6B-C). Comparable results were also obtained in lungs. These data indicate that WNT inhibition interferes with T-cell and macrophage infiltration. Costaining with lead cytokines further highlighted that treatment with WNT inhibitors decreased the numbers of interleukin-4 (IL-4)+ Th2 cells and IL-13+ M2 macrophages in the skin of allografted mice (supplemental Figure 7).
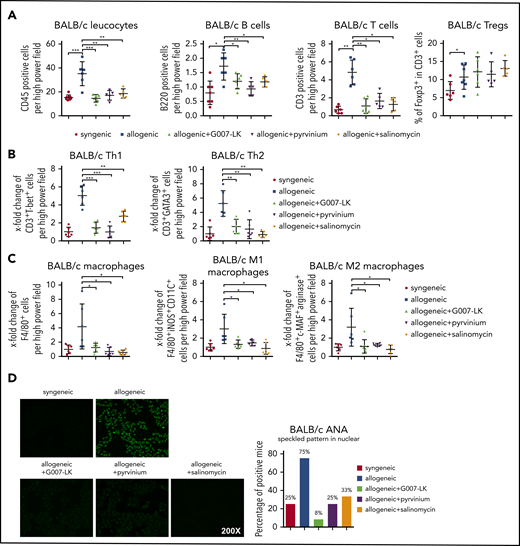
Treatment with the WNT inhibitor G007-LK, pyrvinium, or salinomycin prevents leukocyte infiltration in cGVHD-induced dermal fibrosis in the B10.D2 (H-2d) → BALB/c (H-2d) model. (A) WNT inhibitors effectively prevented the infiltration of leukocytes, particularly B cells and T cells, in murine skin after allogeneic transplantation without changing regulatory T cells. (B) Treatment with WNT inhibitors reduced Th1 and Th2 cell infiltration in murine skin after allogeneic transplantation. (C) WNT inhibitors reduced total macrophage, M1 macrophage, and M2 macrophage infiltration in allogeneic murine skin. (D) Representative immunofluorescence images of HEp-2 cells incubated with murine serum for ANA detection (original magnification ×200; left panel). Percentages of ANA+ mice (n = 6 mice per group; right panel). *P < .5; **P < .05; ***P < .01.
Treatment with the WNT inhibitor G007-LK, pyrvinium, or salinomycin prevents leukocyte infiltration in cGVHD-induced dermal fibrosis in the B10.D2 (H-2d) → BALB/c (H-2d) model. (A) WNT inhibitors effectively prevented the infiltration of leukocytes, particularly B cells and T cells, in murine skin after allogeneic transplantation without changing regulatory T cells. (B) Treatment with WNT inhibitors reduced Th1 and Th2 cell infiltration in murine skin after allogeneic transplantation. (C) WNT inhibitors reduced total macrophage, M1 macrophage, and M2 macrophage infiltration in allogeneic murine skin. (D) Representative immunofluorescence images of HEp-2 cells incubated with murine serum for ANA detection (original magnification ×200; left panel). Percentages of ANA+ mice (n = 6 mice per group; right panel). *P < .5; **P < .05; ***P < .01.
Consistent with the decrease in B-cell counts in fibrotic tissues, we also observed reduced formation of antinuclear antibodies (ANAs) in mice treated with WNT inhibitors. ANAs with speckled pattern were observed in the majority of vehicle-treated B10.D2 (H-2d) → BALB/c (H-2d) mice and LP/J (H-2b) → C57BL/6 (H-2b) mice, but their frequency decreased significantly in mice treated with G007-LK, pyrvinium, or salinomycin (Figure 5D; supplemental Figure 8).
Effects of WNT inhibition on GVL
Given the effects of WNT inhibition on leukocyte infiltration into fibrotic skin, we next aimed to investigate potential effects of WNT inhibition on GVL using the A20 leukemia model of GVL. All BALB/c (H-2d) → BALB/c (H-2d) mice injected with 20 000 A20 cells 10 days after irradiation died of tumor-related complications within 62 days after injection (44.3 ± 12.1 days, mean ± standard deviation [SD]) (Figure 6A). Vehicle-treated B10.D2 (H-2d) → BALB/c (H-2d) mice demonstrated improved survival, with survival periods of up to 88 days (69 ± 13.7 days, mean ± SD). The survival time of G007-LK–, pyrvinium-, or salinomycin-treated B10.D2 (H-2d) → BALB/c (H-2d) mice was significantly longer than that of BALB/c (H-2d) → BALB/c (H-2d) mice, but there was only a small statistically nonsignificant difference compared with vehicle-treated B10.D2 (H-2d) → BALB/c (H-2d) mice. Mice with allogeneic transplantation demonstrated reduced numbers of A20 cells by flow cytometry compared with mice with syngeneic transplantation. The number of A20 tumor cells in mice with allogeneic transplantation was not altered by application of any of the WNT inhibitors at day 11, 16, or 25 post–A20 injection. WNT inhibition also did not affect A20 counts in mice with syngeneic transplantation, excluding a major direct effect on tumor cells as confounder in this model (Figure 6B). These findings may indicate that WNT inhibition preferentially affects type 2 immune responses, with only minor effects on allorecognition and GVL.
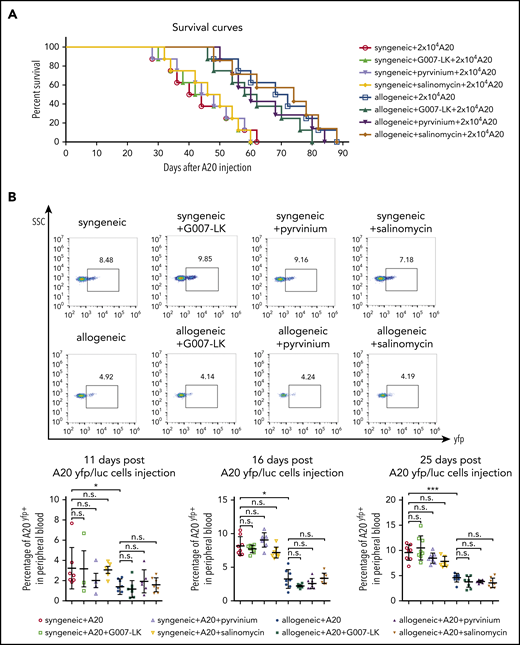
Effects of WNT inhibition on GVL and survival in the A20 leukemia model. (A) In mice injected with 20 000 A20 cells, comparable survival times were noted in G007-LK–, pyrvinium-, salinomycin-, or vehicle-treated mice with syngeneic or allogeneic transplantation. Survival over time is shown as Kaplan-Meier curves. P < .005, allogeneic vs syngeneic. For WNT inhibitor treatment groups, no significant differences compared with vehicle-treated mice (n = 8 mice per group). (B) Percentage of yfp+ tumor cells in the peripheral blood of G007-LK–, pyrvinium-, salinomycin-, or sham-treated mice with syngeneic or allogeneic transplantation injected with A20 yfp/luc cells, as determined by flow cytometry on days 11, 16, and 25 post–A20 injection (n = 8 mice per group). *P < .5; **P < .05; ***P < .01. ns, not significant.
Effects of WNT inhibition on GVL and survival in the A20 leukemia model. (A) In mice injected with 20 000 A20 cells, comparable survival times were noted in G007-LK–, pyrvinium-, salinomycin-, or vehicle-treated mice with syngeneic or allogeneic transplantation. Survival over time is shown as Kaplan-Meier curves. P < .005, allogeneic vs syngeneic. For WNT inhibitor treatment groups, no significant differences compared with vehicle-treated mice (n = 8 mice per group). (B) Percentage of yfp+ tumor cells in the peripheral blood of G007-LK–, pyrvinium-, salinomycin-, or sham-treated mice with syngeneic or allogeneic transplantation injected with A20 yfp/luc cells, as determined by flow cytometry on days 11, 16, and 25 post–A20 injection (n = 8 mice per group). *P < .5; **P < .05; ***P < .01. ns, not significant.
Clinical characteristics of patients with sclGVHD after alloSCT
| Clinical characteristic | Data |
|---|---|
| Patients, N | 12 |
| Sex | |
| Female | 4 (33) |
| Male | 8 (67) |
| Age of recipient (mean ± SD), y | 49 ± 17 |
| Age of donor (mean ± SD), y | 42 ± 9 |
| Time between alloSCT and onset of cGVHD (mean ± SD), d | 250 ± 40 |
| Prior acute GVHD | 8 (75) |
| Clinically active disease | |
| Yes | 12 (100) |
| No | 0 (0) |
| Therapy | |
| CSA + Pred + Rux | 5 (42) |
| CSA + Pred + ECP | 4 (33) |
| CSA + Pred + MMF + ECP | 1 (8) |
| Steroid + CSA | 2 (17) |
| Clinical characteristic | Data |
|---|---|
| Patients, N | 12 |
| Sex | |
| Female | 4 (33) |
| Male | 8 (67) |
| Age of recipient (mean ± SD), y | 49 ± 17 |
| Age of donor (mean ± SD), y | 42 ± 9 |
| Time between alloSCT and onset of cGVHD (mean ± SD), d | 250 ± 40 |
| Prior acute GVHD | 8 (75) |
| Clinically active disease | |
| Yes | 12 (100) |
| No | 0 (0) |
| Therapy | |
| CSA + Pred + Rux | 5 (42) |
| CSA + Pred + ECP | 4 (33) |
| CSA + Pred + MMF + ECP | 1 (8) |
| Steroid + CSA | 2 (17) |
Unless otherwise noted, data are n (%).
CSA, cyclosporine A; ECP, extracorporeal photopheresis; MMF, mycophenolate mofetil; Pred, prednisolone; Rux, ruxolitinib.
Discussion
Our results demonstrate that canonical WNT signaling plays a key role in the pathogenesis of tissue remodeling in sclGVHD after alloSCT. This finding is consistent with the prominent role of WNT signaling in other fibrotic diseases, including systemic sclerosis.48-50 RNASeq highlights a consistent WNT-induced gene expression profile in the skin of patients with sclGVHD compared with controls. Consistently, β-catenin accumulated in fibroblasts of patients with sclGVHD, promoting the transcription of WNT target genes, such as AXIN2. We also observed a prominent canonical WNT signature in human sclGVHD skin. Activation of canonical WNT signaling was not restricted to human sclGVHD; it was also observed in murine sclGVHD. B10.D2 (H-2d) → BALB/c (H-2d) mice and LP/J (H-2b) → C57BL/6 (H-2b) mice also showed accumulation of nuclear β-catenin and increased levels of Axin2 mRNA. Together, these data provide strong evidence for aberrant activation of canonical WNT signaling in sclGVHD. Our data support previous studies highlighting deregulation of several WNT regulators, such as DKK3, secreted frizzled-related protein 4, or R-spondin in cGVHD.34,35,37 In particular, DKK3 showed potential as a biomarker for cGVHD.34
Targeting of canonical WNT signaling inhibited fibroblast-to-myofibroblast transition and collagen deposition in vitro and in 2 models of murine sclGVHD. However, in addition to direct effects on fibroblasts, indirect mechanisms may contribute to the potent inhibition of fibrotic remodeling in vivo. We demonstrate that inhibition of canonical WNT signaling also reduced T-cell, B-cell, and macrophage infiltration. These leukocyte populations are major sources of profibrotic mediators, such as IL-4 and IL-13, with potent activating effects on fibroblasts. Indeed, we found decreased levels of T cells and macrophages positive for IL-4 and IL-13. The molecular mechanisms underlying these anti-inflammatory effects require further studies. In particular, it remains to be elucidated whether the impaired leukocyte counts upon WNT inhibition are caused by direct effects of WNT signaling on leukocytes, impaired release of proinflammatory mediators by fibroblasts, or a combination of both. Consistent with a role for a direct effect of WNT inhibitors on inflammatory cells, canonical WNT signaling has been shown to modulate the phagocytic activity of macrophages and regulate B-cell proliferation, T-cell development, and cytokine release from macrophages.51-61 However, these findings provide evidence that G007-LK, pyrvinium, and salinomycin may target fibrotic tissue remodeling via direct effects on fibroblasts, as well as indirectly by blocking the release of profibrotic mediators from infiltrating leukocytes. This dual mode of action may contribute to the potent antifibrotic effects of WNT inhibition, with almost complete prevention of cGVHD-induced dermal and pulmonary fibrosis.
Of particular note, we demonstrate that WNT inhibition does not ameliorate GVL. G007-LK, pyrvinium, or salinomycin did not significantly affect survival of mice injected with A20 cells after allogeneic transplantation or alter the number of circulating tumor cells. These effects were not mediated by direct inhibitory effects of WNT inhibitors on A20 cells, as shown by treatment of mice with syngeneic transplantation with G007-LK, pyrvinium, or salinomycin. Rather, WNT inhibition may offer the potential to selectively interfere with GVHD-associated pathomechanisms without interfering with the desired GVL effect of allogeneic transplantation.
We demonstrate in 2 mouse models of sclGVHD that inhibition of canonical WNT signaling is an effective treatment for experimental cGVHD. Three compounds with different modes of action, G007-LK, pyrvinium, and salinomycin, all abolish the aberrant activation of canonical WNT signaling in cGVHD. The inhibition of leukocyte accumulation and fibroblast activation by WNT inhibitors translated into potent amelioration of clinical and histological features of sclGVHD. Treatment with G007-LK, pyrvinium, or salinomycin reduced weight loss, improved skin manifestations, such as hyperkeratosis, alopecia, and skin ulcers, and prevented collagen accumulation and myofibroblast differentiation in skin and lungs of allografted mice. It remains to be determined whether those histological changes in the lungs in response to WNT inhibition translate into functional changes, as measured by pulmonary function tests. Our findings are also supported by the effects of niclosamide in murine cGVHD. Although niclosamide is not selective for canonical WNT signaling and targets other fibrosis-relevant pathways, such as STAT3, ERK, AKT, and NOTCH signaling with comparable efficacy, its antifibrotic effects provide further evidence for targeting canonical WNT signaling in sclGVHD.62 Although additional studies to assess the potential of WNT inhibitors to induce regression of established fibrosis are desirable to further characterize the therapeutic potential of WNT inhibitors in sclGVHD, our findings may have translational implications. Pyrvinium is the standard treatment for oxyuriasis, and it has been in clinical use for this indication for decades.63-65 Several tankyrase inhibitors are being evaluated in clinical trials for cancer (http://clinicaltrials.gov; NCT03562832). Salinomycin is an antibiotic that is broadly used in veterinary medicine and was tested against human cancer stem cells.66,67 Other drugs with WNT-inhibitory properties, including porcupine inhibitors, also entered clinical trials (http://clinicaltrials.gov; NCT02649530). Although the clinical experience from approved drugs, such as pyrvinium, and new results from clinical trials with other WNT inhibitors indicate a favorable profile with short-term treatment, management of a chronic disease, such as sclGVHD, would require long-term therapy. Given the role of WNT signaling in stem cell renewal, prolonged use may cause toxicity in tissues with high cell turnover, such as the gut or the bone marrow. This is of particular concern in patients with GVHD; GVHD and the conditioning regimen may have already damaged the respective tissue and their stem cells. Although we did not observe an exacerbation of gastrointestinal symptoms or loss of LGR5+ intestinal stem cells in our mice upon treatment with G007-LK, pyrvinium, or salinomycin in antifibrotic doses, further experiments and careful monitoring are required. In humans with year-long treatment, strategies to minimize toxicity upon long-term application, such as intermittent dosing or combination therapies to lower the required doses of individual drugs, might be required. Topical application might be an option for cutaneous sclGVHD. Indeed, topical application of the WNT inhibitor C-82, which blocks the interaction of CBP with β-catenin, has been evaluated in a proof-of-concept trial in patients with systemic sclerosis.68 Although it failed to show differences in clinical outcomes, modest changes in histological outcomes and effects on the expression of fibrosis-relevant genes were reported.68,69 Inhibition of WNT signaling may also interfere with normal wound healing.
In summary, we demonstrate that canonical WNT signaling plays an important role in the pathophysiology of sclGVHD. Canonical WNT signaling is hyperactive in sclGVHD and promotes fibroblast activation, most likely by a combination of direct and indirect inflammation-related mechanisms. Inhibition of canonical WNT signaling by G007-LK, pyrvinium, or salinomycin, 3 compounds with different modes of action, ameliorates clinical and histological signs of experimental sclGVHD, without significant inhibition of GVL. Because different inhibitors of canonical WNT signaling are in clinical use or are in advanced clinical development, targeting of canonical WNT signaling might be a novel treatment approach for sclGVHD.
Data sharing requests should be sent to Jörg H. W. Distler (joerg.distler@uk-erlangen.de).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Wolfgang Espach and Lena Summa for excellent technical assistance.
This work was supported by grants DI 1537/7-1, DI 1537/8-1, DI 1537/9-1 and -2, DI 1537/11-1, DI 1537/12-1, DI 1537/13-1, DI 1537/14-1, DI 1537/17-1, DE 2414/2-1, DE 2414/4-1 and ZH 809/1-1 from the German Research Foundation; SFB CRC1181 (Project C01) and SFB TR221/Project 324392634 (B04) also from the German Research Foundation; grants A64 and F1-06 from the Interdisziplinäre Zentrum für Klinische Forschung; Interdisciplinary Center for Clinical Research (IZKF) in Erlangen; grant 2013.056.1 from the Wilhelm-Sander-Foundation; grants 2014_A47, 2014_A248, and 2014_A184 from the Else-Kröner-Fresenius-Foundation; grant 14-12-17-1-Bergmann from the ELAN-Foundation Erlangen, BMBF, MASCARA program, TP 2 (Molecular Assessment of Signatures Characterizing the Remission of Arthritis, subproject 2); and a Career Support Award of Medicine from the Ernst Jung Foundation.
Authorship
Contribution: Y.Z., L.S., K.D., T.T.-M., X.M., C.T.-M., C.D., A.-E.M., H.Z., and C.-W.C. performed experiments and/or analyzed data; Y.Z., A.B., W.H., A.M., G.S., B.M.S., and J.H.W.D. designed the research plan and wrote the manuscript; S. Krauss provided inhibitor G007-LK and discussed the manuscript; and M.D., M.Z., J.W., D.W., and S. Karrer contributed skin samples from patients with sclGVHD, discussed the results, and reviewed the final manuscript.
Conflict-of-interest disclosure: J.H.W.D. has acted as a consultant for Actelion, Active Biotech, Anamar, Bayer Pharma, Boehringer Ingelheim, Celgene, Galapagos, GlaxoSmithKline, Inventiva, JB Therapeutics, Medac, Pfizer, Ruiyi, and Union Chimique Belge (UCB); has received research funding from Anamar, Active Biotech, Array Biopharma, aTyr Pharma, Bristol Myers Squibb, Bayer Pharma, Boehringer Ingelheim, Celgene, Galapagos, GlaxoSmithKline, Inventiva, Novartis, Sanofi-Aventis, RedX, and UCB; and owns stock in 4D Science. D.W. has received honoraria from Novartis, Mallinckrodt, Takeda, and Neovi. The remaining authors declare no competing financial interests.
Correspondence: Jörg H. W. Distler, Department of Internal Medicine III–Rheumatology and Immunology, Friedrich-Alexander-University Erlangen-Nürnberg and University Hospital Erlangen, Ulmenweg 18, 91054 Erlangen, Germany; e-mail: joerg.distler@uk-erlangen.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal