


Abstract
VEXAS syndrome (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) is a monogenic disease of adulthood caused by somatic mutations in UBA1 in hematopoietic progenitor cells. Patients develop inflammatory and hematologic symptoms. Myeloid-driven autoinflammation and progressive bone marrow failure lead to substantial morbidity and mortality. Effective medical treatments need to be identified. Reports in the current issue of Blood describe novel UBA1 genetic variants, treatment options, and insight into disease pathophysiology. VEXAS syndrome represents a prototype for a new class of diseases.
Introduction
VEXAS syndrome (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) was first reported in 2020 in 25 men with adult-onset inflammatory disease and myeloid dysplasia.1 Using a genotype-first approach to disease discovery, acquired mutations were identified in all cases of VEXAS in the UBA1 gene, which encodes for the master enzyme of cellular ubiquitylation.2 These mutations were novel (ie, absent from public databases, including the Genome Aggregation Database). The name VEXAS is an acronym based upon key features of the syndrome. Vacuoles are seen in myeloid and erythroid progenitor cells from bone marrow aspirates. E1 enzyme refers to the ubiquitin activating enzyme encoded by UBA1, which is an X-linked gene. Mutations in UBA1 are lineage restricted to myeloid cells and result in autoinflammatory disease. Finally, this disease presents late in life as the result of somatic mutations in blood.3
Clinical description of VEXAS syndrome
VEXAS is a severe, progressive disease with clinical features that bridge rheumatologic and hematologic conditions. Systemic inflammation involving the skin, lungs, blood vessels, and cartilage often leads to the assignment of various clinical diagnoses, including Sweet syndrome, relapsing polychondritis, polyarteritis nodosa, and giant cell arteritis. Additionally, patients with VEXAS suffer from a spectrum of hematologic problems, including macrocytic anemia, thrombocytopenia, thromboembolic disease, and progressive bone marrow failure, which can evolve to hematologic malignancy (Figure 1). An increased risk for hematologic malignancy, most notably myelodysplastic syndrome (MDS), has been reported in many rheumatologic diseases, and conversely, MDS has been associated with a variety of autoimmune syndromes.4-8 VEXAS syndrome may explain some of these historic clinical associations.
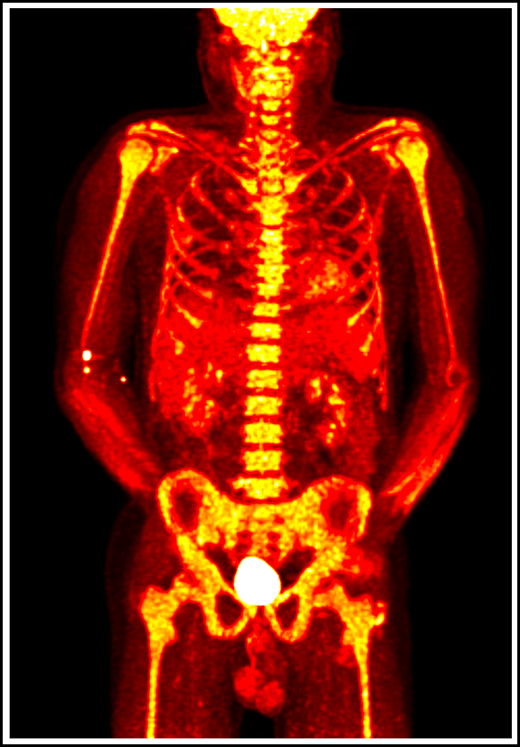
18F-fluorodeoxyglucose positron emission tomography in a patient with VEXAS syndrome demonstrating hypermetabolic activity in bone marrow (yellow). Somatic mutations in UBA1 in hematopoietic cells lead to myeloid-driven inflammation that is often refractory to treatment in patients with VEXAS syndrome.
18F-fluorodeoxyglucose positron emission tomography in a patient with VEXAS syndrome demonstrating hypermetabolic activity in bone marrow (yellow). Somatic mutations in UBA1 in hematopoietic cells lead to myeloid-driven inflammation that is often refractory to treatment in patients with VEXAS syndrome.
A diagnosis of VEXAS should be considered in patients with treatment-refractory inflammatory disease with associated progressive hematologic abnormalities. Among patients with a clinical diagnosis of relapsing polychondritis, male sex plus a mean corpuscular volume >100fL or platelet count <200 ✕ 109/L predicted VEXAS syndrome with near-perfect accuracy.9
Bourbon et al,10 Gurnari et al,11 Lytle et al,12 and Poulter et al13 add to the evolving clinical understanding of the VEXAS syndrome. In aggregate, they detail an additional 24 cases of VEXAS. The identification of a substantial number of patients so quickly after the first report of the syndrome suggests an underappreciated prevalence of this disease. Similar to the initial publication,1 cases were only identified in men with disease onset in the fifth decade of life or later. Systemic inflammation manifested as vasculitis, chondritis, and neutrophilic dermatosis, among other symptoms and signs. The clinical similarities to the initial description of patients with VEXAS may reflect the stereotypical nature of the syndrome or selection bias due to preferential screening of patients with autoinflammatory disease and myeloid dysplasia.
Genetic diagnosis of VEXAS syndrome
UBA1 is an X-linked gene that escapes X inactivation.14 To date, VEXAS has been reported exclusively in men, and women likely are protected by the unmutated allele. In the original description, all 25 men with VEXAS had missense mutations in codon 41 of UBA1.1 These mutations were seen in hematopoietic progenitor cells in bone marrow and lineage restricted to myeloid cells in circulation. Bourbon et al10 and Poulter et al13 describe additional mutations in UBA1 that do not involve codon 41. Two patients had a novel variant in the splice motif at the junction of intron 2 and exon 3 (C.118-1G>C), resulting in a UBA1 protein lacking methionine 41. The authors speculate that this mutation likely leads to the formation of a catalytically inactive cytoplasmic isoform of UBA1, as previously described.1 An additional novel variant (C.167C>T; p.Ser56Phe)13 in a single patient was also reported to be restricted to myeloid cells, leading to temperature-dependent impairment of the resulting isoform.
Vacuoles as a characteristic feature of VEXAS syndrome
Vacuoles are normal and functional intracellular organelles in plants, fungi, and bacteria,15-17 but cytoplasmic vacuoles in mammalian cells can indicate pathology.18 Vacuoles in affected cells can be transient or irreversible, and the latter implies a permanent intrinsic defect.18,19 Vacuoles are rare in marrow myeloid and erythroid precursor cells. Gurnari et al11 screened 11c772 BM samples but found only 24 with cytoplasmic vacuoles. When cytoplasmic vacuoles are identified in these lineages on morphologic examination of marrow for cytopenia, the differential diagnosis includes alcohol intoxication,20 copper deficiency/zinc toxicity,21-23 and myeloid neoplasms.24 Sequencing of UBA1 variants now needs to be included in the evaluation of an adult patient with cytoplasmic vacuoles in the marrow. Vacuoles in myeloid and erythroid precursors have been present in all patients with VEXAS thus far (in the initial report1 and now by Bourbon et al,10 Gurnari et al,11 Lytle et al,12 and Poulter et al13) Cytoplasmic vacuoles are predominantly localized in promyelocytes, myelocytes, erythroid precursors, and blasts in the marrow from VEXAS patients.11 Features that may aid in differentiating VEXAS from other etiologies of myeloid and erythroid precursor cell cytoplasmic vacuolization include presence of autoinflammatory manifestations, macrocytic anemia as a predominant cytopenia followed by thrombocytopenia, cytopathology with numerous coarse vacuoles in both myeloid and erythroid lineages, and normal copper levels. Exactly what vacuoles contain is not clear and needs further investigation.
VEXAS syndrome and hematologic malignancies
The association between autoinflammation and myeloid malignancies is well described in the literature,4,25 but VEXAS establishes a genetic link for the co-occurrence of these heterogenous disorders. MDS has been diagnosed at a high frequency in patients with VEXAS, including 25% (6/25; Beck et al1), 30% (3/10; Poulter et al13), and 55% (6/11; Bourbon et al10). The risk of developing MDS with acquired UBA1 mutation appears to be much higher than observed with well-established clonal hematopoietic disease such as paroxysmal nocturnal hemoglobinuria (2% to 6% by 10 years).26 UBA1 is a key regulator of cellular protein degradation, a pathway not within the current list of genes associated with MDS.27 Whether UBA1 mutation represents a new driver clone for myeloid neoplasm or the occurrence of MDS in VEXAS is driven by other clones selected in chronic inflammatory microenvironment is not known. Two reports identified large DNMT3A clones in one patient each with MDS (43% variant allele frequency [VAF]1 and 24%VAF,11 respectively); smaller clones in MLL-PTD (3.45%), CSF1R (3.12%), and SF3B1 (1%) were present in other MDS patients,1 but their clinical significance is unclear. The molecular landscape in VEXAS MDS is not typical of classical MDS, in which myeloid neoplasia gene mutations are common, large clones are present, and multiple genes are often involved.28,29 Further studies of the clonal genetic landscape in patients with VEXAS will provide insight into the role of inflammation in the pathophysiology of MDS.
In addition to MDS, acquisition of UBA1 mutation predisposes to multiple myeloma (MM) or MM and MDS both. Lytle et al12 presented a case of a 68-year-old male with a history of myeloma and relapsing polychondritis whose bone marrow biopsy, which was performed for progressive pancytopenia, showed features that were diagnostic for both MDS with multilineage dysplasia and residual myeloma. As more patients are identified earlier in the VEXAS disease course, prospective follow-up should include enhanced screening for both of these malignancies.
Treatment of VEXAS syndrome
Understanding the molecular basis of a particular disease is an important first step toward developing more effective treatments. The VEXAS syndrome is associated with considerable morbidity and high mortality. Symptoms are typically refractory to treatment, and high-dose glucocorticoids are only temporizing and have substantial toxicity. Bourbon et al10 and Poulter et al13 emphasize the treatment-refractory nature of VEXAS, as most patients received several steroid-sparing agents in addition to concomitant glucocorticoids. Due to the retrospective study design, Bourbon et al10 evaluated “time to next treatment” as a proxy for effectiveness. The hypomethylating agent azacytidine was used for the longest median duration (21.9 months), but no improvement in cytopenia or myelodysplastic features on bone marrow was observed. Janus kinase inhibitors were effective for some features of systemic inflammatory disease, particularly skin involvement. Prospective evaluation of treatment efficacy is needed to define optimal clinical management. Nuances in clinical phenotype may inform treatment approaches in VEXAS. For example, hypomethylating agents might be preferentially considered in a subset of patients with concomitant MDS. Animal models of VEXAS could enable preclinical research to better understand the pathophysiology of the disease and provide insight into novel therapeutic targets. A zebrafish model, replicating loss of the cytoplasmic isoform of UBA1, demonstrates upregulation of multiple inflammatory cytokines that are elevated in VEXAS (eg, tumor necrosis factor, interleukin-1 [IL-1], IL-6, and IL-8).1 Development of an animal model in a higher organism poses some challenges, as the mutations associated with VEXAS syndrome are likely embryonic lethal. High-risk therapies such as allogeneic bone marrow transplantation should be considered in select patients with VEXAS syndrome given the clonal nature of the disease, persistent and progressive hyperinflammation from complex activation of multiple innate immune pathways, and predisposition to hematologic malignancies.
Future directions
The identification of the VEXAS syndrome serves as another link between clonal hematopoiesis and systemic inflammation.30,31 VEXAS may be a prototype for a new class of “hematoinflammatory diseases.” These diseases would be defined by somatic mutations in hematopoietic cells, systemic inflammation, and the potential to evolve into overt myelodysplastic, myeloproliferative, or lymphoproliferative disease. Acquired mutations in STAT3 in lymphocytes underlie a proportion of patients with Felty syndrome and predispose to large granular lymphocyte leukemia.32 Somatic mutations in BRAF in histiocytes are causal in some patients with Erdheim-Chester disease and may lead to aortitis and myeloproliferative neoplasms.33,34 A series of lymphoma driver mutations transform B cells to produce pathogenic autoantibodies that predispose the development of cryoglobulinemic vasculitis in Sjogren syndrome, a disease associated with increased risk for non-Hodgkin lymphoma.35 In the reports of Bourbon et al10 and Poulter et al,13 8 of 19 patients and 8 of 18 patients, respectively, had myeloid dysplasia and autoinflammation without detectable mutations in UBA1, but further genomic studies may reveal additional novel acquired mutations in UBA1-mutation–negative patients.
Early observations in VEXAS syndrome and related diseases are helping to define the role somatic mutations play in complex, adult-onset diseases33 and provide a framework for collaboration in the clinic and the research laboratory between hematologists and rheumatologists.
Authorship
Contribution: P.C.G., B.A.P., and N.S.Y. designed research, performed research, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Peter C. Grayson, NIAMS/NIH, 10 Center Dr, Building 10, 10N Rm 216G, Bethesda, MD 02892; e-mail: peter.grayson@nih.gov.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal