

Key Points
Addition of ixazomib to Rd in nontransplant NDMM patients resulted in a nonstatistically significant increase in PFS (HR, 0.830; P = .073).
Ixazomib-Rd is a feasible and tolerable all-oral triplet regimen in this setting, with a well-characterized and manageable safety profile.

Abstract
Continuous lenalidomide-dexamethasone (Rd)-based regimens are among the standards of care in transplant-ineligible newly diagnosed multiple myeloma (NDMM) patients. The oral proteasome inhibitor ixazomib is suitable for continuous dosing, with predictable, manageable toxicities. In the double-blind, placebo-controlled TOURMALINE-MM2 trial, transplant-ineligible NDMM patients were randomized to ixazomib 4 mg (n = 351) or placebo (n = 354) plus Rd. After 18 cycles, dexamethasone was discontinued and treatment was continued using reduced-dose ixazomib (3 mg) and lenalidomide (10 mg) until progression/toxicity. The primary endpoint was progression-free survival (PFS). Median PFS was 35.3 vs 21.8 months with ixazomib-Rd vs placebo-Rd, respectively (hazard ratio [HR], 0.830; 95% confidence interval, 0.676-1.018; P = .073; median follow-up, 53.3 and 55.8 months). Complete (26% vs 14%; odds ratio [OR], 2.10; P < .001) and ≥ very good partial response (63% vs 48%; OR, 1.87; P < .001) rates were higher with ixazomib-Rd vs placebo-Rd. In a prespecified high-risk cytogenetics subgroup, median PFS was 23.8 vs 18.0 months (HR, 0.690; P = .019). Overall, treatment-emergent adverse events (TEAEs) were mostly grade 1/2. With ixazomib-Rd vs placebo-Rd, 88% vs 81% of patients experienced grade ≥3 TEAEs, 66% vs 62% serious TEAEs, and 35% vs 27% TEAEs resulting in regimen discontinuation; 8% vs 6% died on study. Addition of ixazomib to Rd was tolerable with no new safety signals and led to a clinically meaningful PFS benefit of 13.5 months. Ixazomib-Rd is a feasible option for certain patients who can benefit from an all-oral triplet combination. This trial was registered at www.clinicaltrials.gov as #NCT01850524.
Introduction
Autologous stem cell transplantation (ASCT) is the standard of care in young (aged ≤65 years) and select fit, elderly patients with newly diagnosed multiple myeloma (NDMM).1,2 However, the median age at diagnosis is ∼69 to 70 years,3,4 and the majority of NDMM patients are ineligible for ASCT due not only to advanced age but also to the presence of comorbidities, such as severe renal or lung impairment, and cardiac and liver disease.5,6 In the newly diagnosed setting, differing treatment options are required due to the diverse patient population, which encompasses fit 70-plus-year-olds as well as elderly and/or frail patients6-10 with poor performance status. Within this heterogeneous population, there are various patient subgroups with an unmet clinical need who experience poorer outcomes, including elderly/frail patients,5,6 patients with high-risk cytogenetics,9-11 and patients with renal impairment12 or bone lesions.13,14
Continuous lenalidomide-dexamethasone (Rd)-based regimens, including Rd alone, daratumumab-Rd, and bortezomib-Rd, are among the standards of care for transplant-ineligible NDMM patients.15-21 Use of proteasome inhibitors (PIs) in a continuous fashion or to higher cumulative doses leads to improved long-term outcomes,22-24 and PI-based triplet regimens show superior outcomes vs non–PI-based doublets in this patient population.19,25 However, long-term administration of injectable PIs may be challenging, and there may be various patients who would benefit from an all-oral PI-Rd triplet, such as patients who are unable to, or prefer not to, travel frequently to the hospital/clinic.
The oral PI ixazomib has demonstrated an efficacy and safety profile amenable to weekly administration at a fixed dose26-28 and can be given for extended periods of time in routine clinical practice with predictable and manageable toxicities.29-31 Ixazomib is approved in the United States and European Union in combination with Rd for the treatment of multiple myeloma (MM) patients who have received ≥1 prior therapy.32-34 Ixazomib-Rd has also been investigated in early-phase trials in NDMM patients, demonstrating rates of very good partial response or better (≥ VGPR) of 58% to 68% and promising progression-free survival (PFS) and overall survival (OS).26,35 We therefore conducted the phase 3, double-blind, placebo-controlled, multicenter TOURMALINE-MM2 study (#NCT01850524), which compared ixazomib-Rd vs placebo-Rd in patients with NDMM who were candidates for Rd treatment but transplant-ineligible due to age or comorbidities.
Patients and methods
Patients
Adult patients with a confirmed diagnosis of symptomatic MM according to International Myeloma Working Group (IMWG) criteria and who were eligible for treatment with Rd but ineligible for ASCT due to age (≥65 years) or comorbidities were enrolled (see supplemental Table 1, available on the Blood Web site, for detailed eligibility criteria). The trial was conducted in accordance with the International Conference on Harmonization Good Clinical Practice guideline and appropriate regulatory requirements. Local ethics committees or institutional review boards approved the protocol. All patients provided written informed consent.
Study design
Patients were randomized (1:1) to receive oral ixazomib 4 mg or matching placebo capsule on days 1, 8, and 15, plus oral lenalidomide 25 mg on days 1 to 21 (10 mg for patients with creatinine clearance [CrCl] ≤60 or ≤50 mL/min, depending on local prescribing information) and oral dexamethasone 40 mg on days 1, 8, 15, and 22 (reduced to 20 mg in patients aged >75 years at randomization) of each dosing cycle in 28-day cycles. Randomization was stratified by age (<75 vs ≥75 years), International Staging System (ISS) disease stage (I or II vs III), and Brief Pain Inventory-Short Form (BPI-SF) worst pain score (<4 vs ≥4) at screening (see supplemental data for details). Patients continued treatment for 18 cycles or until progressive disease (PD) or unacceptable toxicity, whichever came first. After 18 cycles, dexamethasone was discontinued, and patients continued the assigned drug regimen with reduced dose levels of ixazomib 3 mg and lenalidomide 10 mg until progression or unacceptable toxicity. Prophylactic medications, permitted concomitant treatments, and details of dose adjustments for toxicities are listed in the supplemental data.
The primary endpoint was PFS (time from randomization to first documentation of disease progression or death of any cause) as assessed by an independent review committee blinded to both patient treatment assignment and investigator assessment. Prespecified key secondary endpoints were OS, complete response (CR) rate per IMWG criteria, and pain response rate based on BPI-SF score. Other secondary endpoints included overall response rate (ORR), time to response, duration of response, time to progression (TTP), progression-free survival 2 (PFS2) (time from randomization to second disease progression on subsequent line of anticancer therapy or death of any cause), OS, and PFS in patients with an expanded group of high-risk cytogenetic abnormalities [del(17p), amp(1q21), t(4;14), t(14;16)], minimal residual disease (MRD)-negative status, safety, and change in global health status. Evaluation of health care resource utilization (HRU) was a prespecified exploratory endpoint.
Assessments
Response assessments were performed every cycle until disease progression, or every 4 weeks in patients who discontinued treatment prior to disease progression. Response and disease progression assessments were based on central laboratory results and IMWG 2011 criteria.36 Response/progression assessments for PFS2 were performed using local laboratory results at a recommended frequency of once every 12 weeks. All patients were followed for survival after disease progression (every 12 weeks until death or study termination). Cytogenetic abnormalities were assessed by a central laboratory using bone marrow aspirate samples taken at screening. Safety was assessed throughout the study, and treatment-emergent adverse events (TEAEs) were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03. The supplemental data provides details of assessments of pain, cytogenetic abnormalities, MRD, health-related quality of life (HRQoL), and HRU.
Statistical analysis
The study used a closed sequential testing procedure for the primary endpoint of PFS and the key secondary endpoints of OS, CR, and pain response rate, in that order for a family-wise 2-sided level of significance of 5%. Two interim analyses plus a final analysis were planned. The study was adequately powered to test PFS and OS superiority. For PFS, 370 events were needed to provide 92% power assuming a hazard ratio (HR) of 0.70 and 2-sided α .04. For OS superiority, 320 OS events were required to provide 80% power assuming HRs of 0.72 and 2-sided α .05. Significance boundaries for PFS and OS were calculated separately using the γ(−1) and O’Brien-Fleming α-spending functions based on the observed number of events at each analysis. That is, if PFS achieved significance, the OS event reestimation was to be done to increase the required number of events from 320 to a maximum of 400. In the case that PFS achieved significance in the intent-to-treat (ITT) population, OS in the ITT population was planned to be tested using an O’Brien-Fleming stopping boundary for efficacy calculated using a Lan-DeMets α-spending function.37 CR rate was to be tested at the same α level as OS when OS reached significance, and pain response rate was then to be tested at the same α level as that for CR rate when CR rate reached statistical significance. All other efficacy endpoints were evaluated with noninferential analyses (not type I error protected; tested at a 2-sided α level of .05).
The first interim analysis was planned to be performed when ∼326 PFS events had occurred (328 actual). Because the threshold for early stopping for futility or a preemptive declaration of statistical significance was not crossed, per protocol, the study continued in a blinded manner for the final analysis for PFS. This was planned to be performed when ∼370 (378 actual) PFS events had occurred and is reported here. At this analysis, PFS was statistically tested in the ITT population (α = .04) and in 3 prespecified subgroup populations in parallel (total α = .01), using the Hochberg procedure for multiplicity correction. The prespecified subgroups were patients with baseline CrCl >60 mL/min and patients aged <75 years, which were selected based on data suggesting the potential for treatment benefit in these groups plus the poorer outcomes seen with Rd in the opposite subgroups,38,39 and patients with expanded high-risk cytogenetic abnormalities defined as del(17p), t(4;14), t(14;16), and amp(1q21), which was selected based on data indicating the particular benefit of PI-based treatment in this subgroup.40
Analysis populations are defined in the supplemental data. Kaplan-Meier methodology was used to estimate time-to-event distributions, with stratified log-rank tests and Cox models used for interarm comparisons of time-to-event endpoints. Subgroup analyses were conducted for PFS relative to baseline stratification factors, demographic data, and disease characteristics. A stratified Cochran-Mantel-Haenszel χ2 test was used to assess interarm differences in response and pain response rates.
Results
Patients
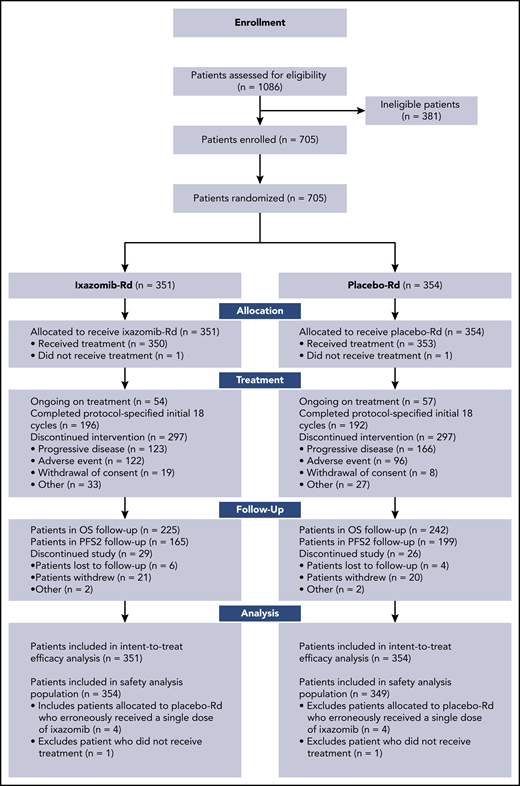
Between May 17, 2013 and December 29, 2015, 705 patients from 157 sites in 8 countries in Europe, North America, and Asia-Pacific were enrolled (Figure 1), 351 patients were assigned to ixazomib-Rd, and 354 patients were assigned to placebo-Rd. Baseline demographic and disease characteristics were well balanced between arms (Table 1). Median age was 73 vs 74 years; 43.0% vs 44.1% of patients were aged ≥75 years; 16.0% vs 16.7% of patients had stage III disease; 53.8% vs 53.7% had a BPI-SF score of ≥4; 38.2% vs 41.2% had expanded high-risk cytogenetic abnormalities, and 57.8% vs 57.6% had CrCl >60 mL/min.
Patient demographics and baseline disease characteristics
| Ixazomib-Rd (N = 351) | Placebo-Rd (N = 354) | |
|---|---|---|
| Median age (range), y | 73 (48-90) | 74 (48-88) |
| Age categories, y, n (%) | ||
| <65 | 11 (3.1) | 8 (2.3) |
| 65 to <75 | 189 (53.8) | 190 (53.7) |
| 75 to <85 | 134 (38.2) | 145 (41.0) |
| ≥85 | 17 (4.8) | 11 (3.1) |
| Sex, n (%) | ||
| Male | 172 (49.0) | 182 (51.4) |
| Female | 179 (51.0) | 172 (48.6) |
| Race, n (%) | ||
| White | 291 (82.9) | 285 (80.5) |
| Asian | 44 (12.5) | 52 (14.7) |
| Black or African American | 11 (3.1) | 13 (3.7) |
| Other | 5 (1.4) | 4 (1.1) |
| ECOG performance status, n (%) | ||
| 0 | 110 (31.3) | 105 (29.7) |
| 1 | 183 (52.1) | 198 (55.9) |
| 2 | 58 (16.5) | 51 (14.4) |
| ISS stage at study entry, n (%) | ||
| I | 171 (48.7) | 153 (43.2) |
| II | 123 (35.0) | 142 (40.1) |
| III | 56 (16.0) | 59 (16.7) |
| Missing | 1 (0.3) | 0 |
| Revised-ISS stage at study entry, n (%) | ||
| I | 107 (30.5) | 96 (27.1) |
| II | 222 (63.2) | 241 (68.1) |
| III | 21 (6.0) | 17 (4.8) |
| Missing | 1 (0.3) | 0 |
| BPI-SF worst pain rating at screening, n (%) | ||
| <4 | 162 (46.2) | 164 (46.3) |
| ≥4 | 189 (53.8) | 190 (53.7) |
| Cytogenetics, n (%)* | ||
| High risk [t(4;14), t(14;16), del(17p)] | 60 (17.1) | 63 (17.8) |
| Corresponding standard risk | 231 (65.8) | 234 (66.1) |
| Unclassifiable for high risk | 60 (17.1) | 57 (16.1) |
| Expanded high risk [t(4;14), t(14;16), del(17p), amp(1q21)] | 134 (38.2) | 146 (41.2) |
| Corresponding standard risk | 155 (44.2) | 139 (39.3) |
| Unclassifiable for expanded high risk | 62 (17.7) | 69 (19.5) |
| CrCl, n (%) | ||
| ≤60 mL/min | 148 (42.2) | 150 (42.4) |
| >60 mL/min | 203 (57.8) | 204 (57.6) |
| Elevated LDH, n (%) | 43 (12.3) | 32 (9.0) |
| Extramedullary disease at study entry, % | 11 (3.1) | 11 (3.1) |
| Ixazomib-Rd (N = 351) | Placebo-Rd (N = 354) | |
|---|---|---|
| Median age (range), y | 73 (48-90) | 74 (48-88) |
| Age categories, y, n (%) | ||
| <65 | 11 (3.1) | 8 (2.3) |
| 65 to <75 | 189 (53.8) | 190 (53.7) |
| 75 to <85 | 134 (38.2) | 145 (41.0) |
| ≥85 | 17 (4.8) | 11 (3.1) |
| Sex, n (%) | ||
| Male | 172 (49.0) | 182 (51.4) |
| Female | 179 (51.0) | 172 (48.6) |
| Race, n (%) | ||
| White | 291 (82.9) | 285 (80.5) |
| Asian | 44 (12.5) | 52 (14.7) |
| Black or African American | 11 (3.1) | 13 (3.7) |
| Other | 5 (1.4) | 4 (1.1) |
| ECOG performance status, n (%) | ||
| 0 | 110 (31.3) | 105 (29.7) |
| 1 | 183 (52.1) | 198 (55.9) |
| 2 | 58 (16.5) | 51 (14.4) |
| ISS stage at study entry, n (%) | ||
| I | 171 (48.7) | 153 (43.2) |
| II | 123 (35.0) | 142 (40.1) |
| III | 56 (16.0) | 59 (16.7) |
| Missing | 1 (0.3) | 0 |
| Revised-ISS stage at study entry, n (%) | ||
| I | 107 (30.5) | 96 (27.1) |
| II | 222 (63.2) | 241 (68.1) |
| III | 21 (6.0) | 17 (4.8) |
| Missing | 1 (0.3) | 0 |
| BPI-SF worst pain rating at screening, n (%) | ||
| <4 | 162 (46.2) | 164 (46.3) |
| ≥4 | 189 (53.8) | 190 (53.7) |
| Cytogenetics, n (%)* | ||
| High risk [t(4;14), t(14;16), del(17p)] | 60 (17.1) | 63 (17.8) |
| Corresponding standard risk | 231 (65.8) | 234 (66.1) |
| Unclassifiable for high risk | 60 (17.1) | 57 (16.1) |
| Expanded high risk [t(4;14), t(14;16), del(17p), amp(1q21)] | 134 (38.2) | 146 (41.2) |
| Corresponding standard risk | 155 (44.2) | 139 (39.3) |
| Unclassifiable for expanded high risk | 62 (17.7) | 69 (19.5) |
| CrCl, n (%) | ||
| ≤60 mL/min | 148 (42.2) | 150 (42.4) |
| >60 mL/min | 203 (57.8) | 204 (57.6) |
| Elevated LDH, n (%) | 43 (12.3) | 32 (9.0) |
| Extramedullary disease at study entry, % | 11 (3.1) | 11 (3.1) |
ECOG, Eastern Cooperative Oncology Group; FISH, fluorescence in situ hybridization; LDH, lactate dehydrogenase.
In accordance with the protocol, the cutoff values for defining the presence of high-risk cytogenetic abnormalities were established by the central diagnostic laboratory on the basis of the false positive rates (or technical cutoff values) of the FISH probes that were used. These cutoff points were 5% positive cells for del(17p), 3% positive cells for t(4;14) and t(14;16), and 20% positive cells for amp(1q21).
At data cutoff (December 2, 2019), 54 (15.4%) and 57 (16.1%) patients were ongoing on therapy in the ixazomib-Rd and placebo-Rd arms, respectively (Figure 1), and study treatment had been discontinued in 297 (84.6%) and 297 (83.9%) patients. The primary reasons for treatment discontinuation were PD in 35.0% and 46.9% of patients and adverse events in 34.8% and 27.1%, respectively (Figure 1).
Efficacy
Median follow-up for PFS was 53.3 and 55.8 months in the ixazomib-Rd and placebo-Rd arms, respectively. With 169 and 209 independent review committee–assessed PFS events in the ixazomib-Rd and placebo-Rd arms, respectively (supplemental Table 2), median PFS was 35.3 vs 21.8 months (HR, 0.830; 95% CI, 0.676-1.018; P = .073) (Figure 2A).
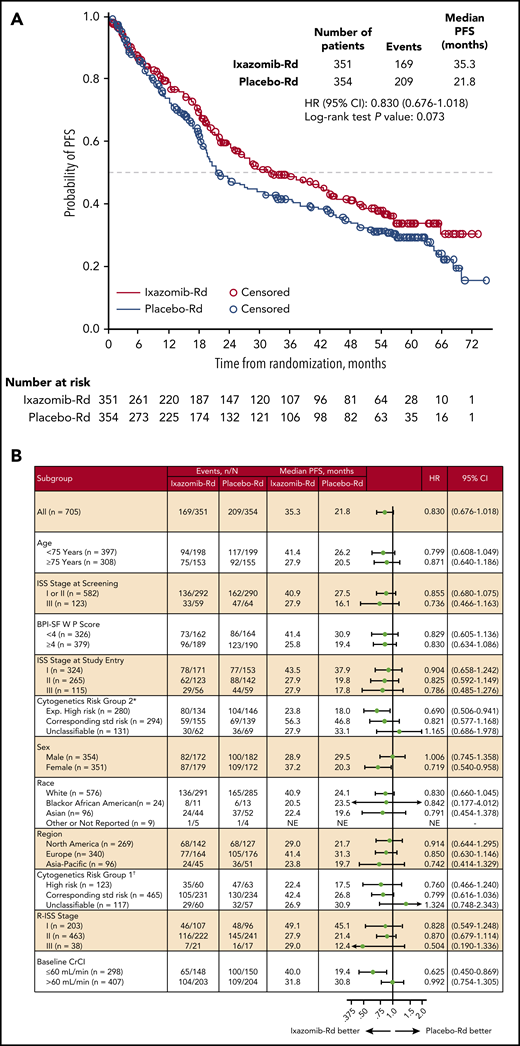
PFS by independent review of the ITT population. (A) Kaplan-Meier analysis of PFS by independent review of the ITT population. (B) Forest plots of PFS in prespecified subgroups based on patient and disease characteristics of the ITT population. *Expanded high-risk category includes del(17p), t(4;14), t(14;16), and amp(1q21) abnormalities; standard (std) risk category includes normal results as well as all types of abnormalities other than t(4;14), t(14;16), del(17), or amp(1q21); unclassifiable for expanded high risk is defined as patients who do not have cytogenetic data that can be categorized to expanded high risk or std risk corresponding to expanded high-risk group, because of either missing, unknown or indeterminate results. †High-risk category includes del(17p), t(4;14), or t(14;16) abnormalities; std risk category includes normal results as well as all types of abnormalities other than t(4;14), t(14;16), or del(17); unclassifiable for high risk is defined as patients who do not have cytogenetic data that can be categorized to high risk or std risk corresponding to high risk group, either because of missing, unknown, or indeterminate results. BPI-SF W P, Brief Pain Inventory-Short Form worst pain; CI, confidence interval; Exp., expanded; NE, not evaluable; R-ISS, revised ISS.
PFS by independent review of the ITT population. (A) Kaplan-Meier analysis of PFS by independent review of the ITT population. (B) Forest plots of PFS in prespecified subgroups based on patient and disease characteristics of the ITT population. *Expanded high-risk category includes del(17p), t(4;14), t(14;16), and amp(1q21) abnormalities; standard (std) risk category includes normal results as well as all types of abnormalities other than t(4;14), t(14;16), del(17), or amp(1q21); unclassifiable for expanded high risk is defined as patients who do not have cytogenetic data that can be categorized to expanded high risk or std risk corresponding to expanded high-risk group, because of either missing, unknown or indeterminate results. †High-risk category includes del(17p), t(4;14), or t(14;16) abnormalities; std risk category includes normal results as well as all types of abnormalities other than t(4;14), t(14;16), or del(17); unclassifiable for high risk is defined as patients who do not have cytogenetic data that can be categorized to high risk or std risk corresponding to high risk group, either because of missing, unknown, or indeterminate results. BPI-SF W P, Brief Pain Inventory-Short Form worst pain; CI, confidence interval; Exp., expanded; NE, not evaluable; R-ISS, revised ISS.
In the 3 prespecified subgroups for which the study was designed for statistical testing in parallel, median PFS was 23.8 vs 18.0 months (HR, 0.690; 95% CI, 0.506-0.941; P = .019) in patients with expanded high-risk cytogenetics, 41.4 vs 26.2 months (HR, 0.799; 95% CI, 0.608-1.049; P = .106) in patients aged <75 years, and 31.8 vs 30.8 months (HR, 0.992; 95% CI, 0.754-1.305; P = .955) in patients with CrCl > 60 mL/min (supplemental Figure 1). Figure 2B shows PFS with ixazomib-Rd vs placebo-Rd in all other prespecified subgroups based on patient and disease characteristics, including in patients with ISS stage III disease at screening (HR, 0.736; 95% CI, 0.466-1.163) and those with CrCl ≤60 mL/min (HR, 0.625; 95% CI, 0.450-0.869).
CR rates were 25.6% vs 14.1% in patients in the ixazomib-Rd vs placebo-Rd arms (P < .001), and rates of ≥ VGPR were 63.0% and 47.7%, respectively (P < .001) (Table 2). Median time to response was 1.0 (95% CI, 0.99-1.08) vs 1.9 months (95% CI, 1.15-1.87) in the ixazomib-Rd vs placebo-Rd arms (HR, 1.402; 95% CI, 1.185-1.659; P < .001). Among 287 and 281 evaluable responding patients in the ixazomib-Rd and placebo-Rd arms, 112 (39.0%) and 141 (50.2%) had relapsed/progressed or died at data cutoff; median duration of response was 50.6 (95% CI, 39.98-not estimable) vs 37.5 (95% CI, 25.69-50.27) months.
Best confirmed responses, pain responses, and MRD evaluation in the ITT population
| Ixazomib-Rd (N = 351), n (%) (exact 95% CI) | Placebo-Rd (N = 354), n (%) (exact 95% CI) | OR (95% CI) | P | |
|---|---|---|---|---|
| Confirmed best response | ||||
| CR including sCR | 90 (25.6) (21.2-30.5) | 50 (14.1) (10.7-18.2) | 2.10 (1.43-3.09) | <.001 |
| VGPR | 131 (37.3) (32.2-42.6) | 119 (33.6) (28.7-38.8) | ||
| CR + VGPR (including sCR) | 221 (63.0) (57.7-68.0) | 169 (47.7) (42.4-53.1) | 1.87 (1.38-2.53) | <.001 |
| PR | 67 (19.1) (15.1-23.6) | 113 (31.9) (27.1-37.1) | ||
| ORR (CR + PR + VGPR [including sCR]) | 288 (82.1) (77.6-85.9) | 282 (79.7) (75.1-83.7) | 1.16 (0.79-1.70) | .436 |
| SD | 31 (8.8) (6.1-12.3) | 37 (10.5) (7.5-14.1) | ||
| PD | 4 (1.1) (0.3-2.9) | 14 (4.0) (2.2-6.5) | ||
| Not evaluable | 28 (8.0) (5.4-11.3) | 21 (5.9) (3.7-8.9) | ||
| Evaluation of MRD by flow cytometry (sensitivity 10−5) | ||||
| Patients evaluated for MRD status | 101 (28.8) | 62 (17.5) | ||
| Patients who were MRD-negative, n/N (%) | 53/101 (52.5) | 24/62 (38.7) | ||
| Pain response | ||||
| BPI-SF worst pain score ≥4 at baseline* | 190 (54.1) | 195 (55.1) | ||
| Pain response,† n/N (%) | 96/190 (50.5) (43.2-57.8) | 100/195 (51.3) (44.0-58.5) | 0.98 (0.66-1.46) | .920 |
| Ixazomib-Rd (N = 351), n (%) (exact 95% CI) | Placebo-Rd (N = 354), n (%) (exact 95% CI) | OR (95% CI) | P | |
|---|---|---|---|---|
| Confirmed best response | ||||
| CR including sCR | 90 (25.6) (21.2-30.5) | 50 (14.1) (10.7-18.2) | 2.10 (1.43-3.09) | <.001 |
| VGPR | 131 (37.3) (32.2-42.6) | 119 (33.6) (28.7-38.8) | ||
| CR + VGPR (including sCR) | 221 (63.0) (57.7-68.0) | 169 (47.7) (42.4-53.1) | 1.87 (1.38-2.53) | <.001 |
| PR | 67 (19.1) (15.1-23.6) | 113 (31.9) (27.1-37.1) | ||
| ORR (CR + PR + VGPR [including sCR]) | 288 (82.1) (77.6-85.9) | 282 (79.7) (75.1-83.7) | 1.16 (0.79-1.70) | .436 |
| SD | 31 (8.8) (6.1-12.3) | 37 (10.5) (7.5-14.1) | ||
| PD | 4 (1.1) (0.3-2.9) | 14 (4.0) (2.2-6.5) | ||
| Not evaluable | 28 (8.0) (5.4-11.3) | 21 (5.9) (3.7-8.9) | ||
| Evaluation of MRD by flow cytometry (sensitivity 10−5) | ||||
| Patients evaluated for MRD status | 101 (28.8) | 62 (17.5) | ||
| Patients who were MRD-negative, n/N (%) | 53/101 (52.5) | 24/62 (38.7) | ||
| Pain response | ||||
| BPI-SF worst pain score ≥4 at baseline* | 190 (54.1) | 195 (55.1) | ||
| Pain response,† n/N (%) | 96/190 (50.5) (43.2-57.8) | 100/195 (51.3) (44.0-58.5) | 0.98 (0.66-1.46) | .920 |
PR, partial response; sCR, stringent complete response; SD, stable disease.
BPI-SF baseline data differ from data at screening used for stratification, as the BPI is a patient-reported tool in which the patient reports the pain they are currently experiencing and have experienced within the previous 24 h.
Pain response is defined as the occurrence of at least a 30% reduction from baseline in BPI-SF worst pain score over the last 24 h without an increase in analgesic use for 2 consecutive measurements ≥28 d apart.
Bone marrow aspirates were taken from 28.8% and 17.5% of patients in the ixazomib-Rd and placebo-Rd arms, respectively, for MRD evaluation. Of these patients, 52.5% and 38.7% were MRD-negative by flow cytometry (Table 2), corresponding to overall MRD-negative rates in the ITT population of 15.1% and 6.8%, respectively. Among 190 and 195 patients in the ixazomib-Rd and placebo-Rd arms with a worst pain score of ≥4 at baseline, pain response rates were 50.5% and 51.3%, respectively (Table 2).
The median TTP was 45.8 vs 26.8 months (HR, 0.738; 95% CI, 0.589-0.925; P = .008) with ixazomib-Rd vs placebo-Rd (supplemental Figure 2). The median TTP was 49.3 vs 30.9 months (HR, 0.767; 95% CI, 0.572-1.030) in patients aged <75 years, and 43.5 vs 23.6 months (HR, 0.697; 95% CI, 0.490-0.994) in patients aged ≥75 years (supplemental Figure 2). Median PFS2 was 63.2 vs 52.2 months (HR, 0.859; 95% CI, 0.684-1.078; P = .189) (supplemental Figure 3). Median OS was not reached in either arm (HR, 0.998; 95% CI, 0.790-1.261) after a median follow-up of ∼58 months, and with 284 patients having died, 136 and 148 patients in the ixazomib-Rd and placebo-Rd arms, respectively (Figure 3). Of these patients, 64 died prior to disease progression, of whom 27 (18 vs 9 patients receiving ixazomib-Rd vs placebo-Rd) died within the first 6 months of randomization (supplemental Table 3). The remaining 37 patients (19 vs 18 receiving ixazomib-Rd vs placebo-Rd) died after 6 months of randomization.
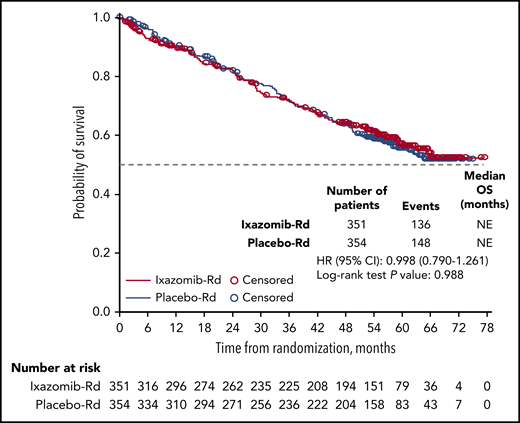
Kaplan-Meier analysis of OS by independent review on the ITT population. OS distributions in the ixazomib-Rd and placebo-Rd arms.
Kaplan-Meier analysis of OS by independent review on the ITT population. OS distributions in the ixazomib-Rd and placebo-Rd arms.
Overall, 46.6% vs 57.0% of patients in the ixazomib-Rd vs placebo-Rd arms, respectively, had progressed to second-line therapy at the time of analysis. Of these, 70.9% vs 85.9%, respectively, received a PI-containing therapy. Median OS for patients who received a PI-containing therapy was 54.3 vs 57.0 months (HR, 1.124; 95% CI, 0.803-1.573; P = .495), and for those who had not, median OS was not reached in the ixazomib-Rd arm and 51.8 months in the placebo-Rd arm (HR, 0.524; 95% CI, 0.243-1.131; P = .095).
Treatment exposure and safety
The safety population included 354 and 349 patients in the ixazomib-Rd and placebo-Rd arms, respectively (Figure 1). Patients received a median of 20 treatment cycles in each arm, with 55.4% and 55.0% of patients having completed the initial 18 cycles of treatment in the ixazomib-Rd and placebo-Rd arms, respectively (60.6% vs 59.5% aged <75 years; 48.3% vs 49.4% aged ≥75 years). Overall, 53.7% and 54.2% of patients in the ixazomib-Rd and placebo-Rd arms, respectively, entered the second phase of treatment with continued ixazomib/placebo plus lenalidomide therapy. Mean relative dose intensities for all agents were slightly lower with ixazomib-Rd, overall, and during cycles 1 to 18 and cycles >18 (Table 3).
Treatment exposure and overall safety profile in the safety population
| Ixazomib-Rd (N = 354) | Placebo-Rd (N = 349) | |
|---|---|---|
| Median number of treatment cycles received, n (range) | 20 (1-80) | 20 (1-81) |
| Completed protocol-specified initial 18 cycles, n (%) | 196 (55.4) | 192 (55.0) |
| Median number of treatment cycles received in patients continuing treatment beyond cycle 18, n (range) | 44.0 (18-80) | 39.0 (19-81) |
| Mean (SD) relative dose intensity,* % | ||
| Ixazomib/placebo | 91.7 (12.5) | 94.8 (9.7) |
| Cycles 1 to 18 | 91.5 (12.9) | 94.9 (10.0) |
| Cycles >18 | 94.6 (7.7) | 96.9 (6.3) |
| Lenalidomide | 80.4 (24.7) | 84.6 (22.9) |
| Cycles 1 to 18 | 81.0 (28.1) | 85.3 (26.2) |
| Cycles >18 | 84.0 (20.2) | 88.0 (20.3) |
| Dexamethasone | 83.2 (20.9) | 86.4 (18.9) |
| Any TEAE, n (%) | 354 (100) | 349 (100) |
| Any drug-related TEAE, n (%) | 342 (96.6) | 323 (92.6) |
| Any grade ≥3 TEAE, n (%) | 312 (88.1) | 284 (81.4) |
| Any drug-related grade ≥3 TEAE, n (%) | 251 (70.9) | 199 (57.0) |
| Any serious TEAE, n (%) | 233 (65.8) | 218 (62.5) |
| Any drug-related serious TEAE, n (%) | 136 (38.4) | 106 (30.4) |
| TEAE resulting in dose reduction of ≥1 of the 3 agents in the study drug regimen, n (%) | 211 (59.6) | 189 (54.2) |
| TEAE resulting in discontinuation of ≥1 of the 3 agents in the study drug regimen, n (%) | 160 (45.2) | 108 (30.9) |
| TEAE resulting in dose discontinuation of the full study drug regimen, n (%) | 124 (35.0) | 94 (26.9) |
| On-study deaths, n (%) | 27 (7.6) | 22 (6.3) |
| Ixazomib-Rd (N = 354) | Placebo-Rd (N = 349) | |
|---|---|---|
| Median number of treatment cycles received, n (range) | 20 (1-80) | 20 (1-81) |
| Completed protocol-specified initial 18 cycles, n (%) | 196 (55.4) | 192 (55.0) |
| Median number of treatment cycles received in patients continuing treatment beyond cycle 18, n (range) | 44.0 (18-80) | 39.0 (19-81) |
| Mean (SD) relative dose intensity,* % | ||
| Ixazomib/placebo | 91.7 (12.5) | 94.8 (9.7) |
| Cycles 1 to 18 | 91.5 (12.9) | 94.9 (10.0) |
| Cycles >18 | 94.6 (7.7) | 96.9 (6.3) |
| Lenalidomide | 80.4 (24.7) | 84.6 (22.9) |
| Cycles 1 to 18 | 81.0 (28.1) | 85.3 (26.2) |
| Cycles >18 | 84.0 (20.2) | 88.0 (20.3) |
| Dexamethasone | 83.2 (20.9) | 86.4 (18.9) |
| Any TEAE, n (%) | 354 (100) | 349 (100) |
| Any drug-related TEAE, n (%) | 342 (96.6) | 323 (92.6) |
| Any grade ≥3 TEAE, n (%) | 312 (88.1) | 284 (81.4) |
| Any drug-related grade ≥3 TEAE, n (%) | 251 (70.9) | 199 (57.0) |
| Any serious TEAE, n (%) | 233 (65.8) | 218 (62.5) |
| Any drug-related serious TEAE, n (%) | 136 (38.4) | 106 (30.4) |
| TEAE resulting in dose reduction of ≥1 of the 3 agents in the study drug regimen, n (%) | 211 (59.6) | 189 (54.2) |
| TEAE resulting in discontinuation of ≥1 of the 3 agents in the study drug regimen, n (%) | 160 (45.2) | 108 (30.9) |
| TEAE resulting in dose discontinuation of the full study drug regimen, n (%) | 124 (35.0) | 94 (26.9) |
| On-study deaths, n (%) | 27 (7.6) | 22 (6.3) |
SD, standard deviation.
Relative dose intensity defined as: 100 × (total dose received in mg)/(sum of prescribed dose of all treated cycles) for the specified period, for which total prescribed dose equals (dose prescribed × number of prescribed doses per cycle × the number of treated cycles).
For ixazomib/placebo: dose prescribed is 4 mg for the first 18 cycles and 3 mg after cycle 18; number of prescribed doses per cycle is 3.
For lenalidomide: dose prescribed is 25 mg for the first 18 cycles and 10 mg after cycle 18; for patients with baseline CrCl ≤60 mL/min, dose prescribed is 10 mg throughout the study; number of prescribed doses per cycle is 21.
For dexamethasone: dose prescribed is 20 mg for patients >75 y old and 40 mg for all other patients; number of prescribed doses per cycle is 4.
Safety profiles are summarized in Table 3. Overall, 88.1% vs 81.4% of patients in the ixazomib-Rd and placebo-Rd arms, respectively, had grade ≥3 TEAEs; 65.8% vs 62.5% had serious TEAEs; 35.0% vs 26.9% had TEAEs resulting in discontinuation of the full regimen; and 7.6% vs 6.3% of patients died on study. TEAEs were mostly grade 1 or 2; the most common and clinically important any-grade and grade ≥3 TEAEs with ixazomib-Rd and placebo-Rd are summarized in Table 4. Grade ≥3 TEAEs with a ≥5% rate difference between ixazomib-Rd and placebo-Rd included neutropenia (16.9% vs 26.9%), rash (16.7% vs 7.4%), thrombocytopenia (13.3% vs 4.6%), and diarrhea (9.9% vs 2.0%); smaller rate differences were seen for cardiac arrhythmias (10.5% vs 7.4%), pneumonia (10.2% vs 7.4%), and heart failure (4.2% vs 2.9%). There was no difference between arms in the rate of new primary malignancies (Table 4). The incidences of new-onset TEAEs (per Table 3) during cycles 1 to 18 and postcycle 18, and rates of any-grade and grade ≥3 TEAEs with ixazomib-lenalidomide and placebo-lenalidomide postcycle 18, are summarized in supplemental Tables 4 and 5, respectively. Among patients reporting the common any-grade and grade ≥3 TEAEs and TEAEs of clinical importance shown in Table 4, in general, the majority reported their first occurrence of each TEAE within the first 6 months of treatment and, more commonly, within the first 3 months vs after 3 to 6 months (supplemental Table 6).
Most common any-grade (reported in ≥30% of patients on ixazomib-Rd arm) and grade ≥3 TEAEs (reported in ≥10% of patients on ixazomib-Rd arm) plus rates of additional TEAEs of clinical importance, reported with ixazomib-Rd and placebo-Rd in the safety population
| MedDRA preferred term, n (%) | Ixazomib-Rd (N = 354) | Placebo-Rd (N = 349) | ||
|---|---|---|---|---|
| Any grade | Grade ≥3 | Any grade | Grade ≥3 | |
| Diarrhea | 216 (61.0) | 35 (9.9) | 161 (46.1) | 7 (2.0) |
| Rash* | 199 (56.2) | 59 (16.7) | 130 (37.2) | 26 (7.4) |
| Peripheral edema | 172 (48.6) | 4 (1.1) | 117 (33.5) | 4 (1.1) |
| Constipation | 151 (42.7) | 4 (1.1) | 144 (41.3) | 3 (0.9) |
| Nausea | 131 (37.0) | 5 (1.4) | 97 (27.8) | 1 (0.3) |
| Peripheral neuropathy* | 120 (33.9) | 8 (2.3) | 96 (27.5) | 4 (1.1) |
| Fatigue | 109 (30.8) | 14 (4.0) | 106 (30.4) | 14 (4.0) |
| Anemia | 108 (30.5) | 56 (15.8) | 108 (30.9) | 62 (17.8) |
| Vomiting | 105 (29.7) | 4 (1.1) | 46 (13.2) | 2 (0.6) |
| Cardiac arrhythmias*,† | 81 (22.9) | 37 (10.5) | 74 (21.2) | 26 (7.4) |
| Thrombocytopenia* | 73 (20.6) | 47 (13.3) | 33 (9.5) | 16 (4.6) |
| Neutropenia* | 71 (20.1) | 60 (16.9) | 104 (29.8) | 94 (26.9) |
| Pneumonia | 62 (17.5) | 36 (10.2) | 46 (13.2) | 26 (7.4) |
| Acute renal failure* | 58 (16.4) | 23 (6.5) | 65 (18.6) | 26 (7.4) |
| Hypotension* | 41 (11.6) | 8 (2.3) | 29 (8.3) | 7 (2.0) |
| Heart failure*,‡ | 32 (9.0) | 15 (4.2) | 21 (6.0) | 10 (2.9) |
| Liver impairment*,§ | 31 (8.8) | 9 (2.5) | 27 (7.7) | 9 (2.6) |
| Myocardial infarction*,‖ | 11 (3.1) | 6 (1.7) | 9 (2.6) | 8 (2.3) |
| Encephalopathy*,§ | 8 (2.3) | 3 (0.8) | 7 (2.0) | 4 (1.1) |
| New primary malignancies | 43 (12.1) | 40 (11.5) | ||
| MedDRA preferred term, n (%) | Ixazomib-Rd (N = 354) | Placebo-Rd (N = 349) | ||
|---|---|---|---|---|
| Any grade | Grade ≥3 | Any grade | Grade ≥3 | |
| Diarrhea | 216 (61.0) | 35 (9.9) | 161 (46.1) | 7 (2.0) |
| Rash* | 199 (56.2) | 59 (16.7) | 130 (37.2) | 26 (7.4) |
| Peripheral edema | 172 (48.6) | 4 (1.1) | 117 (33.5) | 4 (1.1) |
| Constipation | 151 (42.7) | 4 (1.1) | 144 (41.3) | 3 (0.9) |
| Nausea | 131 (37.0) | 5 (1.4) | 97 (27.8) | 1 (0.3) |
| Peripheral neuropathy* | 120 (33.9) | 8 (2.3) | 96 (27.5) | 4 (1.1) |
| Fatigue | 109 (30.8) | 14 (4.0) | 106 (30.4) | 14 (4.0) |
| Anemia | 108 (30.5) | 56 (15.8) | 108 (30.9) | 62 (17.8) |
| Vomiting | 105 (29.7) | 4 (1.1) | 46 (13.2) | 2 (0.6) |
| Cardiac arrhythmias*,† | 81 (22.9) | 37 (10.5) | 74 (21.2) | 26 (7.4) |
| Thrombocytopenia* | 73 (20.6) | 47 (13.3) | 33 (9.5) | 16 (4.6) |
| Neutropenia* | 71 (20.1) | 60 (16.9) | 104 (29.8) | 94 (26.9) |
| Pneumonia | 62 (17.5) | 36 (10.2) | 46 (13.2) | 26 (7.4) |
| Acute renal failure* | 58 (16.4) | 23 (6.5) | 65 (18.6) | 26 (7.4) |
| Hypotension* | 41 (11.6) | 8 (2.3) | 29 (8.3) | 7 (2.0) |
| Heart failure*,‡ | 32 (9.0) | 15 (4.2) | 21 (6.0) | 10 (2.9) |
| Liver impairment*,§ | 31 (8.8) | 9 (2.5) | 27 (7.7) | 9 (2.6) |
| Myocardial infarction*,‖ | 11 (3.1) | 6 (1.7) | 9 (2.6) | 8 (2.3) |
| Encephalopathy*,§ | 8 (2.3) | 3 (0.8) | 7 (2.0) | 4 (1.1) |
| New primary malignancies | 43 (12.1) | 40 (11.5) | ||
MedDRA, Medical Dictionary for Regulatory Activities; SMQ, standardized MedDRA query.
Higher-level term, SMQ, or pooled term incorporating multiple preferred terms. “Rash” included the preferred terms of rash maculopapular, rash macular, pruritus, rash, rash erythematous, rash popular, pruritus generalized, urticaria, drug eruption, rash pruritic, dermatitis acneiform, purpura, dermatitis allergic, rash generalized, erythema multiforme, rash vesicular, rash morbilliform, Stevens-Johnson syndrome, exfoliative rash, rash follicular, toxic epidermal necrolysis, rash pustular. “Peripheral neuropathy” included the preferred terms of peripheral sensory neuropathy, neuropathy peripheral, peripheral sensorimotor neuropathy, peripheral motor neuropathy. “Cardiac arrhythmias” included the preferred terms of syncope, atrial fibrillation, palpitations, sinus tachycardia, bradycardia, tachycardia, atrioventricular block complete, cardiac arrest, atrial flutter, supraventricular tachycardia, loss of consciousness, sudden death, sinus bradycardia, ventricular extrasystoles, atrioventricular block, arrhythmia, heart rate irregular, bundle branch block right, supraventricular extrasystoles, atrioventricular block first degree, extrasystoles, heart rate increased, sinus node dysfunction, bundle branch block left, electrocardiogram QT prolonged, ventricular tachycardia, cardiorespiratory arrest, heart rate decreased. “Thrombocytopenia” included the preferred terms of thrombocytopenia, platelet count decreased. “Neutropenia” included the preferred terms of neutropenia, neutrophil count decreased. “Acute renal failure” included the preferred terms of blood creatinine increased, acute kidney injury, renal failure, renal impairment, creatinine renal clearance decreased, oliguria, azotemia, nephritis, glomerular filtration rate decreased, proteinuria, renal tubular disorder. “Hypotension” included the preferred terms of hypotension, orthostatic hypotension, anaphylactic reaction. “Heart failure” included the preferred terms of cardiac failure, pulmonary edema, cardiac failure congestive, cardiomegaly, diastolic dysfunction, orthopnea, acute pulmonary edema, pulmonary congestion, right ventricular failure, left ventricular failure. “Liver impairment” included the preferred terms of alanine aminotransferase increased, hypoalbuminemia, aspartate aminotransferase increased, hepatocellular injury, blood alkaline phosphatase increased, γ-glutamyltransferase increased, hyperbilirubinemia, hepatic steatosis, liver function test increased, drug-induced liver injury, hepatic cirrhosis, hepatic function abnormal, cholestasis, hepatic encephalopathy, hepatic enzyme increased, blood bilirubin increased, ascites, hepatitis cholestatic, liver disorder. “Myocardial infarction” included the preferred terms of acute coronary syndrome, angina unstable, acute myocardial infarction, blood creatine phosphokinase increased, coronary artery occlusion, electrocardiogram ST segment elevation, myocardial infarction, troponin increased. “Encephalopathy” included the preferred terms of delirium, hepatic encephalopathy, leukoencephalopathy, encephalopathy, hypoxic-ischemic encephalopathy, posterior reversible encephalopathy syndrome.
Includes 7 (2.0%) and 1 (0.3%) patients with grade 5 events in the ixazomib-Rd and placebo-Rd arms, respectively.
Includes 0 and 1 (0.3%) patients with grade 5 events in the ixazomib-Rd and placebo-Rd arms, respectively.
Includes 1 (0.3%) and 0 patients with grade 5 events in the ixazomib-Rd and placebo-Rd arms, respectively.
Includes 1 (0.3%) and 1 (0.3%) patients with grade 5 events in the ixazomib-Rd and placebo-Rd arms, respectively.
The most common TEAEs resulting in dose reductions of ≥1 agent in the ixazomib-Rd and placebo-Rd arms are summarized in supplemental Table 7. The incidences of individual TEAEs resulting in discontinuation of ≥1 agent or the full regimen were all ≤3% in both arms (supplemental Table 7). Among patients who had a TEAE resulting in discontinuation of ≥1 of the agents in the regimen, 125 of 162 events (77.2%) in the ixazomib-Rd arm and 82 of 109 (75.2%) in the placebo-Rd arm occurred within cycles 1 to 18 (supplemental Table 4), of which 66 of 162 (40.7%) and 33 of 109 (30.3%) occurred within ∼3 months (90 days) (supplemental Table 6).
On-study deaths were considered to be treatment-related in 5 of 27 patients in the ixazomib-Rd arm (sudden death, n = 2; cardiac arrest, lung infection, sepsis, each n = 1), and 4 of 22 patients in the placebo-Rd arm (septic shock, general physical health deterioration, thrombosis, multiple organ dysfunction syndrome, each n = 1).
Mean global health status scores over time indicated similar patient-reported HRQoL in the ixazomib-Rd and placebo-Rd arms (supplemental Figure 4).
HRU during treatment is summarized in supplemental Table 8. Addition of ixazomib to Rd did not result in additional HRU (hospitalizations, emergency room stays, and outpatient visits).
Discussion
The phase 3 TOURMALINE-MM2 study demonstrated that addition of ixazomib to Rd results in a clinically meaningful PFS benefit41 in transplant-ineligible patients with NDMM, with a 13.5-month improvement in median PFS; however, this was not a statistically significant outcome. Efficacy findings also showed improvements in median TTP of 18 to 20 months in the ITT population, and in patients aged <75 and ≥75 years. There was also improvement in CR/≥ VGPR rates with ixazomib-Rd vs placebo-Rd (25.6%/63.0% vs 14.1%/47.7%). The marked difference in TTP and PFS benefit with ixazomib-Rd vs placebo-Rd was possibly driven by the imbalance between arms in the rate of early deaths (within 6 months) in the absence of disease progression, two-thirds of which occurred in the ixazomib-Rd arm. These early death events were investigated for any trends in the cause or patient demographics, and there was no discernible consistent pattern of relatedness to the study drug regimen or of events induced by the disease under study, as assessed by the investigator. For the remaining 37 PFS events attributable to death, deaths were well balanced between the ixazomib-Rd and placebo-Rd regimens (19 and 18, respectively).
Trends in PFS benefit with ixazomib-Rd were seen in patient subgroups, particularly in patients with ISS stage III disease (HR, 0.736), aged <75 years (HR, 0.799), and with CrCl ≤60 mL/min (HR, 0.625). Notably, analysis of the 3 prespecified subgroups statistically tested in parallel demonstrated that ixazomib-Rd improved the poor prognosis of patients with expanded high-risk cytogenetics vs placebo-Rd (HR, 0.690; ITT HR, 0.830), reflecting findings from the TOURMALINE-MM1 trial of ixazomib-Rd vs placebo-Rd in relapsed/refractory MM (HR, 0.664; ITT HR, 0.74).27,40 This appears to be in line with the 2016 IMWG recommendations for treating patients with high-risk cytogenetics with a PI-immunomodulatory drug-dexamethasone triplet combination.11 Further evaluation of the impact of ixazomib in patients with expanded high-risk cytogenetic abnormalities is warranted.
In TOURMALINE-MM2, dexamethasone was discontinued, and ixazomib and lenalidomide doses were notably reduced from cycle 19 onwards (∼60% reduction in lenalidomide dose), the impact of which is discernible in the shape of the PFS curves, in which the rates of PFS events increased at ∼18 months. Supporting this, in the FIRST trial, in which patients either received Rd until PD (Rd continuous) or discontinued Rd after 18 months (Rd18), the effect of Rd discontinuation was discernible in the shape of the PFS curves, with the rate of PFS events being greater after ∼18 months in the Rd18 arm vs Rd continuous arm.18 The benefit of the prolonged PI approach in TOURMALINE-MM2 is demonstrated by the sustained separation of the PFS curves. In contrast, the absence of OS benefit may be partly explained by the higher number of early deaths unrelated to myeloma in the ixazomib-Rd arm, and partly by differences in subsequent therapies received, which were selected by the investigator. Of those who received subsequent therapy, a higher proportion received a PI in the placebo-Rd arm vs the ixazomib-Rd arm, which resulted in OS slightly favoring placebo-Rd (54.3 vs 57.0 months) in patients who received a PI as next-line therapy. In contrast, for patients receiving non-PI treatment as next-line therapy, OS favored ixazomib-Rd (NE vs 51.8 months). Although these data suggest that PIs as next-line therapy were not as beneficial after progression on ixazomib-Rd compared with placebo-Rd, the overall median PFS2 was 63.2 vs 52.2 months, with a HR of 0.859 in favor of ixazomib-Rd, suggesting that imbalances in third-line treatment and beyond may in part be driving the similarity in OS. Furthermore, advances in salvage therapy since the start of TOURMALINE-MM2, for example, daratumumab-based triplets that were approved in 2016 to 2017, warrant further investigation of the impact of subsequent therapy. More generally, such analyses will contribute to the consideration of optimal therapeutic sequencing in the evolving therapeutic landscape in NDMM and the relapsed setting.
TOURMALINE-MM2 PFS results appear consistent with data from other phase 3 trials of PI-Rd triplet regimens in nontransplant NDMM patients, acknowledging that direct comparisons between studies are confounded by multiple factors, including duration of therapy and study population. Notably, the median PFS with ixazomib-Rd (35.3 months) is very similar to data reported from the ENDURANCE trial of bortezomib-Rd vs carfilzomib-Rd in NDMM patients (34.4 vs 34.6 months), which excluded patients with high-risk cytogenetics [del(17p), t(14;16), t(14;20)]. Patients’ median age was higher in TOURMALINE-MM2 vs ENDURANCE (73 vs 65 years), and actual median duration of ixazomib-Rd treatment in TOURMALINE-MM2 was longer (20 cycles/∼19 months) than that of bortezomib-Rd/carfilzomib-Rd in ENDURANCE (5.9 cycles/8.2 months), albeit the durations of treatment specified per the trial protocols were different. Despite these differences in patient populations and trial designs, comparable PFS results were achieved in these 2 studies of PI-Rd triplets.42 Longer median PFS was reported with bortezomib-Rd vs Rd (40.8 vs 29.0 months; HR, 0.742) in an updated analysis of the SWOG S0777 trial, along with higher ≥ VGPR (74.9%) but similar CR (24.2%) rates; however, again the median age was lower in SWOG S0777 vs TOURMALINE-MM2 (<65 years), and 69% of patients in the bortezomib-Rd arm had an intent to proceed subsequent to ASCT.43 In TOURMALINE-MM2, more than half the patients completed the initial 18 cycles of PI-based triplet therapy. In contrast, in SWOG S0777, only 55.7% of patients received bortezomib-Rd for the protocol-specified maximum of 8 cycles.43 Based on these data in different patient populations, the median PFS achieved with ixazomib-Rd in TOURMALINE-MM2 is clinically meaningful, and the increased treatment duration achieved as a result of the tolerability provides a rationale for this continuous PI-Rd approach in this elderly population. Furthermore, this approach resulted in improved rates of CR/VGPR vs placebo-Rd, although the ORR was similar between arms due to more PRs with placebo-Rd. Thus, the higher rates of deeper response in the ixazomib-Rd arm were reflected in a PFS benefit, but this benefit was not statistically significant compared with the placebo at the 4% level of significance associated with this test.
No new safety signals were seen with ixazomib-Rd. Safety data showed that rates of grade ≥3 TEAEs and TEAEs leading to dose reduction or discontinuation of ≥1 of the regimen drugs were slightly more frequent with ixazomib-Rd vs placebo-Rd, whereas on-study deaths were similar between arms. Overall, rates of TEAEs appeared similar between arms. Rates of grade ≥3 rash, thrombocytopenia, and diarrhea were ≥5% higher in the ixazomib-Rd arm; however, these were manageable toxicities and are consistent with data from prior phase 3 trials of ixazomib.27,28 By contrast, the rate of grade ≥3 peripheral neuropathy in both arms was low (2.3%). Findings from SWOG S0777 and ENDURANCE show that bortezomib-Rd is associated with an elevated risk of grade ≥3 neurologic toxicity, whereas carfilzomib-Rd is associated with higher rates of cardiopulmonary and renal toxicity vs bortezomib-Rd.19,42,43 The tolerable safety profile of ixazomib-Rd in TOURMALINE-MM2 was reflected in similar HRU and patient-reported QoL data between arms. QoL and HRU were maintained from study entry in both arms and were generally similar throughout the study, indicating that ixazomib-Rd did not negatively affect patient-reported QoL vs placebo-Rd. Data from TOURMALINE-MM2 indicate that long-term triplet combinations with oral ixazomib as a PI backbone would be feasible for nontransplant NDMM patients.
A limitation of the present study was that the primary PFS analysis was based on a stratified log-rank test. In addition, use of attenuated treatment after cycle 18 limits interpretation of TOURMALINE-MM2 findings in the context of other studies. Treatment attenuation may partly explain the shorter median PFS in the placebo-Rd arm (21.8 months) vs that with continuous Rd in FIRST (26.0 months), in which patients could continue Rd at the starting doses until disease progression, if tolerated.18 Conversely, median PFS with Rd for 18 cycles in FIRST was 21.0 months,18 whereas median PFS in the EMN01 and RV-MM-PI-0752 trials, in which patients received Rd for only 9 cycles followed by lenalidomide maintenance, was 18.644 and ∼18 to 19 months,45 respectively. Collectively, these data suggest that median PFS in TOURMALINE-MM2 might have been marginally prolonged by continuing full-dose ixazomib and lenalidomide, and/or dexamethasone, for longer; however, at the time of study design, the tolerability of prolonged PI-based triplet therapy was unclear.
In conclusion, the results suggest a clinically meaningful PFS benefit, with a HR of 0.83 and a 13.5-month increase in median PFS, with addition of ixazomib to Rd in transplant-ineligible NDMM patients, but this was not statistically significant. We note that a HR in a range from 1.018, indicating a small negative association, to 0.676, indicating a substantial positive association, is reasonably compatible with our data.41 A trend in PFS benefit was also observed with ixazomib-Rd in patients with expanded high-risk cytogenetics. The ixazomib-Rd safety profile was generally consistent with the well-characterized toxicity profile of ixazomib and ixazomib-Rd in earlier studies,27,28,46 and there was no adverse impact on patient-reported HRQoL in this elderly, transplant-ineligible patient population. The transplant-ineligible NDMM patient population is highly diverse, comprising patients of widely differing fitness and age who often have limited mobility and reduced ability to frequently attend clinic or hospital. Thus, in addition to current regimens requiring regular parenteral administration,16,42,43 novel, tolerable all-oral regimens suitable for extended dosing are important for lessening patients’ treatment burden. TOURMALINE-MM2 demonstrates that ixazomib-Rd is a feasible treatment option for certain transplant-ineligible patients with NDMM who could benefit from an all-oral triplet regimen.
The datasets, including the redacted study protocol, redacted statistical analysis plan, and individual participants’ data supporting the results reported in this article, will be available 3 months from initial request to researchers who provide a methodologically sound proposal. The data will be provided after its deidentification, in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
There is a Blood Commentary on this article in this issue.
Acknowledgments
The authors thank the patients and their families, as well as the TOURMALINE-MM2 study group, the physicians, nurses, study coordinators, and research staff for participation in the trial; Renda Ferrari, of Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, for editorial assistance, and Philippa Lloyd and Steve Hill, of FireKite, an Ashfield company, part of UDG Healthcare plc, for professional medical writing assistance, which was funded by Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, in compliance with Good Publication Practice 3 ethical guidelines.
This study was supported by Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Authorship
Contribution: T.F., S.L., G.Y., P.G.R., and S.V.R. designed the study; T.F., N.J.B., S.-S.Y., P.R., K.S., H.S., L.K., D.J.W., P.M., G.Y., R.M.R., S.R., P.G.R., L.B., S.V.R., and S.K.K. were the study investigators; T.F., N.J.B., S.-S.Y., P.R., E.V., K.S., H.S., F.O., L.K., D.J.W., P.M., R.M.R., S.R., C.P.V., P.G.R., L.B., S.V.R., and S.K.K. enrolled patients; T.F., N.J.B., S.-S.Y., D.J.W., S.L., G.Y., X.Z., and P.T.-A. analyzed the data; T.F., N.J.B., S.-S.Y., K.S., D.J.W., P.M., S.L., G.Y., P.T.-A., R.M.R., C.P.V., P.G.R., L.B., S.V.R., and S.K.K. interpreted the data; all authors prepared, reviewed, and revised the manuscript; and all authors had final approval of the manuscript.
Conflict-of-interest disclosure: T.F. served in an advisory role for Takeda, Roche, Amgen, Celgene, a Bristol Myers Squibb Company, Janssen, Karyopharm, Oncopeptides, and Sanofi; served on speakers bureaus for Takeda, Celgene, a Bristol Myers Squibb Company, and Janssen. C.P.V. received honoraria from Takeda, Amgen, Celgene, a Bristol Myers Squibb Company, Johnson & Johnson, GSK, Janssen, and Sanofi. N.J.B. received grants and personal fees from Celgene, a Bristol Myers Squibb Company, and Janssen; served as consultancy and received honoraria from Takeda, Amgen, Karyopham, Sanofi, GSK, Genentech, and AbbVie. D.J.W. received honoraria from Takeda, Amgen, Antengene, BMS, GSK, Karyopharm, Janssen, and Sanofi. L.K. received honoraria from Amgen, Janssen, Celgene, a Bristol Myers Squibb Company, Sanofi, Takeda, and AbbVie; travel support from Janssen, Amgen, and Takeda; served on advisory boards for Amgen, Janssen, Celgene, a Bristol Myers Squibb Company, and Takeda. S.R. served as a data monitoring board member for Takeda. S.-S.Y. served in a consulting or advisory role for Takeda, Amgen, Astellas, Celgene, a Bristol Myers Squibb Company, Chugai, Janssen, Novartis; received honoraria from Novartis and Amgen; received research funding from Kyowa Kirin, Roche-Genentech, Yuhan Pharmaceutical. K.S. received honoraria from Takeda, Celgene, a Bristol Myers Squibb Company, ONO, Amgen, Novartis, Sanofi, BMS, AbbVie, and Janssen; served in a consulting or advisory role for Takeda, Amgen, Janssen, and Celgene, a Bristol Myers Squibb Company; received research funding from BMS, Celgene, a Bristol Myers Squibb Company, and Amgen. H.S. served on advisory boards, received honoraria and research funding from Chugai and Eisai; served on advisory boards and received research funding from Celgene, a Bristol Myers Squibb Company; served on advisory boards and received honoraria from AstraZeneca; received research funding and honoraria from Takeda, Ono, Nippon Shinyaku, AbbVie, Novartis, Janssen, Sumitomo Dainippon; received research funding from Astellas, Teijin, MSD, Shionogi, Taiho; received honoraria from Kyowa Kirin, Daiichi Sankyo, Fujimoto, Sanofi, BMS, Pfizer, Otsuka, Mundipharma. X.Z. is currently employed by Takeda; P.T.-A. is currently employed by Takeda; G.Y. has been employed in the past by Takeda; R.M.R. has stock ownership and is currently employed at McKesson; has served as consulting or in an advisory role for Takeda, Amgen, Celgene, a Bristol Myers Squibb Company, BMS, Mylan, Coherus BioSciences, Fresenius; was an investigator in AbbVie-sponsored clinical trials; served on an advisory board for Takeda, Amgen, Celgene, a Bristol Myers Squibb Company. P.M. has served as consultancy and received honoraria from Celgene, a Bristol Myers Squibb Company, Janssen, Amgen, Sanofi, AbbVie; received honoraria from Novartis and Takeda. S.L. received personal fees from Takeda, Celgene, a Bristol Myers Squibb Company, BMS, Novartis, Amgen, GSK, AbbVie, Janssen, Merck; served as consultancy from AbbVie, JUNO Therapeutics; served on the board of directors for TG Therapeutics; was employed by Emory University. S.K.K. received research funding, served as consultancy and on advisory boards for Takeda, Celgene, a Bristol Myers Squibb Company, AbbVie, Janssen, Amgen, Genentech/Roche; received research grants from Novartis, Carsgen, Tenebio, Sanofi, and MedImmune; served as consultancy and received research funding from Merck, BMS, and Kite Pharma; served as consultancy for Adaptive Biotechnologies, Karyopharm, and Genecentrix; received other from Cellectar; served on an independent review committee for Oncopeptides; received honoraria from Reddy’s laboratories. P.G.R. served as an advisory committee member for Karyopharm, Oncopeptides, Celgene, a Bristol Myers Squibb Company, Takeda, Janssen, Sanofi, SecuraBio, GSK; received research support from Oncopeptides, Celgene, a Bristol Myers Squibb Company, Takeda. The remaining authors declare no competing financial interests.
Correspondence: Thierry Facon, Centre Hospitalier Universitaire (CHU) Lille, Service des Maladies du Sang, University of Lille, CHRU rue Michel Polonovski, 59037 Lille, France; e-mail: thierry.facon@chru-lille.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal