

Key Points
GGCX mutations differentially affect the carboxylation of vitamin K–dependent proteins associated with distinct clinical phenotypes.
Some GGCX missense mutations affect the pre–messenger RNA splicing rather than altering the corresponding amino acid residues.

Abstract
γ-Glutamyl carboxylase (GGCX) is an integral membrane protein that catalyzes posttranslational carboxylation of a number of vitamin K–dependent (VKD) proteins involved in a wide variety of physiologic processes, including blood coagulation, vascular calcification, and bone metabolism. Naturally occurring GGCX mutations are associated with multiple distinct clinical phenotypes. However, the genotype–phenotype correlation of GGCX remains elusive. Here, we systematically examined the effect of all naturally occurring GGCX mutations on the carboxylation of 3 structure–function distinct VKD proteins in a cellular environment. GGCX mutations were transiently introduced into GGCX-deficient human embryonic kidney 293 cells stably expressing chimeric coagulation factor, matrix Gla protein (MGP), or osteocalcin as VKD reporter proteins, and then the carboxylation efficiency of these reporter proteins was evaluated. Our results show that GGCX mutations differentially affect the carboxylation of these reporter proteins and the efficiency of using vitamin K as a cofactor. Carboxylation of these reporter proteins by a C-terminal truncation mutation (R704X) implies that GGCX’s C terminus plays a critical role in the binding of osteocalcin but not in the binding of coagulation factors and MGP. This has been confirmed by probing the protein–protein interaction between GGCX and its protein substrates in live cells using bimolecular fluorescence complementation and chemical cross-linking assays. Additionally, using a minigene splicing assay, we demonstrated that several GGCX missense mutations affect GGCX’s pre–messenger RNA splicing rather than altering the corresponding amino acid residues. Results from this study interpreted the correlation of GGCX’s genotype and its clinical phenotypes and clarified why vitamin K administration rectified bleeding disorders but not nonbleeding disorders.
Introduction
γ-Glutamyl carboxylase (GGCX), also known as vitamin K–dependent (VKD) carboxylase, is an integral membrane protein residing in the endoplasmic reticulum (ER).1,2 It catalyzes the posttranslational modification of specific glutamate residues to γ-carboxyglutamate (Gla) residues in a number of VKD proteins. Native protein substrates for GGCX include several coagulation factors, as well as osteocalcin (or bone Gla protein [BGP]), matrix Gla protein (MGP), and recently discovered Gla proteins such as Gla-rich protein (GRP) and Gas6.3-5 Carboxylation is essential for the biological function of VKD proteins that control blood coagulation, vascular calcification, bone metabolism, signal transduction, and cancer cell proliferation.6-10
VKD carboxylation was originally observed in clotting factors, and carboxylation of coagulation factors has been extensively studied.11-13 Defects in VKD carboxylation are mainly linked to bleeding disorders, known as combined vitamin K–dependent coagulation factors deficiency (VKCFD).14,15 Comorbid phenotypes have been observed in VKCFD patients that include cardiac, dermatologic, or skeletal abnormalities resulting from insufficient carboxylation of other VKD proteins, such as MGP and/or BGP.16-19 The importance of VKD carboxylation of MGP is supported by the significant associations of cardiovascular diseases and arterial calcifications found in patients taking vitamin K antagonists20 and in free-living people with low dietary menaquinone intakes.21 In addition, in vivo mutagenesis experiments clearly showed that the anti-mineralization function of MGP is dependent on its Gla residues.22 MGP knockout mice died within 2 months because of extensive arterial calcification that led to blood–vessel rupture.23 In humans, defects in MGP carboxylation are also associated with skeletal abnormalities, such as Keutel syndrome.24,25 Additionally, patients with dermatologic abnormalities (eg, pseudoxanthoma elasticum [PXE]-like syndrome) seem to have increased accumulation of uncarboxylated MGP and BGP.26 Furthermore, the carboxylation status of several recently discovered VKD proteins, such as Gla-rich proteins27,28 and proline-rich Gla proteins,29 have been associated with vascular calcification, inflammation, and neurologic disorders.5,30-33
Genetic screenings of patients with vitamin K–related disorders have identified more than 40 naturally occurring mutations in GGCX.17,34,35 Clinical manifestations of these mutations have been classified into 5 distinct phenotypes: the bleeding phenotype (VKCFD) and the nonbleeding phenotypes of cardiac, dermatologic, ophthalmologic, and osseous symptoms.17 VKCFD results from insufficient carboxylation of VKD coagulation factors in the liver and can be ameliorated through the administration of high doses of vitamin K.15 However, nonbleeding phenotypes, caused by defects in carboxylation of extrahepatic Gla proteins, seem to be more difficult to rescue and require much higher doses of vitamin K.25,36 Importantly, some GGCX-associated nonbleeding phenotypes (eg, osseous symptoms) overlap clinically with other syndromes, such as the fetal warfarin syndrome (an embryo disorder resulting from fetal exposure to maternal ingestion of warfarin during pregnancy).37-39 In these cases, GGCX genetic testing is essential for the diagnosis and possible prevention of these irreversible pathologic changes in early life. Recently, De Vilder et al17 systematically explored GGCX genotype–phenotype correlations, reporting that different clinical phenotypes associate with GGCX mutations located at certain protein domains. However, it is not clear why some GGCX mutations cause bleeding disorders, whereas others cause nonbleeding syndromes.
To experimentally clarify the correlation between GGCX genotypes and their clinical phenotypes, we recently established a cell-based system to study GGCX’s function in its native milieu.25 As a proof-of-principle, we clarified the clinical consequences of novel GGCX mutations identified from a patient with a severe cerebral bleeding disorder and with comorbid Keutel syndrome.25 Here, we extended our study to explore the effect of all naturally occurring GGCX mutations on the carboxylation of various VKD proteins (coagulation factors, MGP, and BGP) associated with distinct clinical phenotypes. Our result, that 1 GGCX mutation can differentially affect different VKD proteins’ carboxylation, explains the diverse clinical phenotypes associated with a single GGCX mutation.
Methods
DNA manipulations and plasmid constructions
The GGCX mutations were created by QuickChange site-directed mutagenesis using wild-type GGCX in pBudCE4.1-Met.Luc vector as the template, as previously described.25 Metridia luciferase was used as the internal control for normalizing the variation of transfection efficiency. Chimeric VKD reporter proteins were created by overlap polymerase chain reaction (PCR) and then cloned into the mammalian expression vector pcDNA3.1. Three chimeric reporter-proteins, FIXgla-PC (protein C with its Gla domain exchanged with that of FIX), MGP-sfGFP-His (MGP fused to the N terminus of a superfolder green fluorescent protein with a 6× His tag at the C terminus), and BGP-MBP-His (BGP fused to the N terminus of maltose binding protein with a 8× His-tag at the C terminus), were used to examine the carboxylation of coagulation factors, MGP, and osteocalcin. Fused tags were designed for enzyme-linked immunosorbent assay (ELISA) detection and standard protein purification. For clarity, only VKD proteins (FIXgla-PC, MGP, and BGP) were mentioned in the main text. All other chimeric proteins used in this study were created by overlap PCR, as described elsewhere. The nucleotide sequences of all constructs were verified by DNA sequencing at Eton Bioscience Inc. (Research Triangle Park, NC).
Reporter protein carboxylation in HEK293 cells
The efficiency of reporter protein carboxylation was determined in HEK293 cells with their endogenous GGCX knocked out by clusters of regularly interspaced short palindromic repeats (CRISPR)-Cas9, as previously described with slight modifications.25 Briefly, pcDNA3.1 plasmid containing the cDNA of various chimeric reporter proteins was stably expressed in GGCX-deficient HEK293 cells to obtain reporter cell lines for FIXgla-PC, MGP, and BGP. Then, wild-type GGCX or its mutants were transiently expressed in these cells for evaluating the carboxylation efficiency of different reporter proteins by ELISA.40 To determine the carboxylation efficiency of BGP, a rabbit anti-MBP polyclonal antibody (Proteintech Group, Inc., Rosemont, IL) was used as the capture antibody and a mouse anti-carboxylated BGP (Thermo Fisher Scientific, Waltham, MA) was used as the detection antibody. To determine the carboxylation efficiency of MGP, a rabbit anti-His Tag polyclonal antibody (Proteintech Group, Inc.) was used as the capture antibody and a mouse anti-carboxylated MGP monoclonal antibody (IDS, Boldon, UK)41 was used as the detection antibody. Experimental data were analyzed using GraphPad Prism 8. Statistical analysis was performed using the unpaired Student t test.
Bimolecular fluorescence complementation assay
A split-Venus based bimolecular fluorescence complementation (BiFC) assay was used to study the interaction between GGCX and its protein substrate. This assay is based on the reconstitution of an intact Venus fluorescent protein when 2 of its complementary nonfluorescent fragments are brought together by a pair of interacting proteins in live cells.42,43 The Venus fluorescent protein was divided into 2 fragments: a large fragment containing residues 1 to 173 (VN) and a small fragment containing residues 156 to 238 (VC) (a gift from Andrzej Dziembowski; Addgene plasmid 105804).44 The 2 fragments were fused to GGCX or its VKD protein substrate using overlap PCR. Complementary pairs of the BiFC assay were cotransfected into GGCX-deficient HEK293 cells. To stabilize GGCX and its substrate complex, 5 µM warfarin was included in the cell culture medium. Fluorescence intensity was measured at Ex515nm/Em530nm using a Perkin Elmer Enspire MLD 2300 microplate reader.
GGCX epoxidase activity assay
Cell-based GGCX epoxidase activity was performed in HEK293 cells, as previously described.45 Briefly, wild-type GGCX or its mutants were transiently expressed in GGCX-deficient HEK293 cells. Transfected cells were incubated with cell culture medium containing 5 µM warfarin for 40 hours. Then, the cells were incubated with 20 µM vitamin K and 5 µM warfarin for 6 hours before the K vitamins were extracted from cells for the conventional reversed-phase high-performance liquid chromatography assay.46
Minigene splicing assay
The effect of the GGCX mutation on its pre–messenger RNA (mRNA) splicing was assessed using a minigene splicing assay based on the exon trapping vector pSPL3 (kindly provided by Karen E. Heath, Hospital Universitario La Paz, Spain), as previously described.47,48 Briefly, the affected exons along with their upstream and downstream intronic sequences (∼100 bp on each side) were amplified by PCR using human genomic DNA as the template; the specific primers are listed in supplemental Table 1, available on the Blood Web site. The PCR amplicons were subcloned into the XhoI and BamHI sites of the pSPL3 vector. The minigene constructs were transiently transcribed into HEK293 cells, and the total RNA was extracted for reverse transcriptase-PCR amplification. The PCR products were separated using agarose gel electrophoresis or cloned into a TA cloning vector for DNA sequencing.
Results
Cell-based GGCX functional study using structurally distinct VKD reporter proteins
To better understand the mechanistic differences of GGCX modifying its structurally distinct protein substrates, we selected 3 VKD proteins as reporter proteins (Figure 1A): a chimeric coagulation factor (FIXgla-PC),40 MGP, and BGP. Coagulation factors and MGP bind to GGCX mainly through a tight-binding propeptide,49-51 which is proteolytically removed from the coagulation factors but not from MGP, to form a mature protein after carboxylation. Although the propeptide of BGP has an undetectable affinity to GGCX, BGP binds to GGCX at a site different from the propeptide-recognition site.52 Importantly, the distribution of Gla residues being carboxylated in these proteins is totally different. The Gla residues of coagulation factors are concentrated at the N terminus of the mature protein, immediately preceded by the propeptide (Figure 1A). Unlike coagulation factors, the Gla residues of MGP are scattered on both the N terminus and C terminus of the propeptide sequence, whereas BGP’s Gla residues are located between the 2 GGCX binding sequences.53
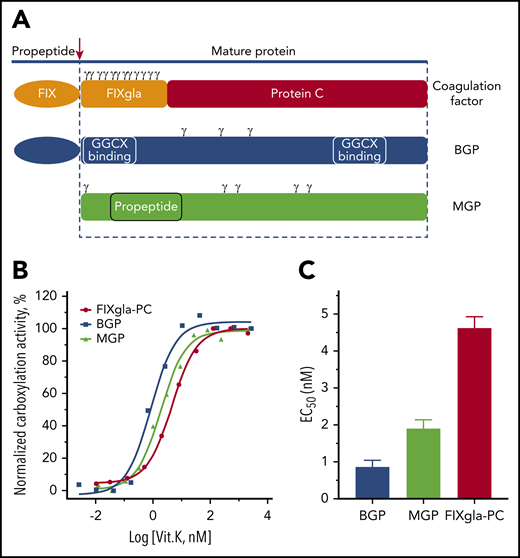
Functional study of GGCX in HEK293 cells using different VKD reporter proteins. (A) Diagram of structurally distinct VKD reporter proteins. Gla residues are indicated by γ. (B) Carboxylation of the 3 VKD reporter proteins in HEK293 cells in response to vitamin K. Wild-type GGCX was transiently expressed in GGCX-deficient HEK293 cells stably expressing FIXgla-PC, MGP, or BGP. Transfected cells were incubated with increasing concentrations of vitamin K for 48 hours. The carboxylation efficiency of the reporter protein was determined in the cell culture medium by ELISA. (C) Half-maximal stimulation concentrations of vitamin K (EC50) (obtained by nonlinear regression using GraphPad Prism 8.0) for the carboxylation of BGP, MGP, and FIXgla-PC. Data are presented as means ± standard deviation (SD).
Functional study of GGCX in HEK293 cells using different VKD reporter proteins. (A) Diagram of structurally distinct VKD reporter proteins. Gla residues are indicated by γ. (B) Carboxylation of the 3 VKD reporter proteins in HEK293 cells in response to vitamin K. Wild-type GGCX was transiently expressed in GGCX-deficient HEK293 cells stably expressing FIXgla-PC, MGP, or BGP. Transfected cells were incubated with increasing concentrations of vitamin K for 48 hours. The carboxylation efficiency of the reporter protein was determined in the cell culture medium by ELISA. (C) Half-maximal stimulation concentrations of vitamin K (EC50) (obtained by nonlinear regression using GraphPad Prism 8.0) for the carboxylation of BGP, MGP, and FIXgla-PC. Data are presented as means ± standard deviation (SD).
We stably expressed these 3 reporter proteins individually in HEK293 cells with their endogenous GGCX knocked out by CRISPR-Cas9.25 To evaluate the 3 reporter cell lines for GGCX’s functional study, we transiently expressed wild-type GGCX in these cells and incubated the transfected cells with increasing concentrations of vitamin K. Results in Figure 1B show that these reporter proteins can be readily carboxylated by GGCX in a vitamin K–dependent manner. It has been shown that glutamate carboxylation and vitamin K epoxidation by GGCX are coupled with a 1:1 stoichiometry.54,55 As there are 3, 5, and 12 glutamate residues in BGP, MGP, and FIXgla-PC, respectively, one would expect that the carboxylation of 1 mole of BGP, MGP, and FIXgla-PC would require 3, 5, and 12 moles of reduced vitamin K, respectively. Our results show that the half-maximal stimulation concentrations (EC50 or apparent Km) of vitamin K for the carboxylation of BGP, MGP, and FIXgla-PC are 0.86, 1.9, and 4.6 nM, respectively (Figure 1C). These EC50 values correlate well to the number of glutamate residues in the reporter proteins, suggesting that the established reporter cell lines are good tools for the functional study of GGCX’s carboxylation of different VKD proteins.
GGCX mutations differentially affect carboxylation of the 3 reporter proteins
Currently, more than 40 GGCX mutations have been identified from patients with vitamin K–related disorders.17,34,35 These mutations are scattered throughout the GGCX molecule (Figure 2A). To explore how these mutations affect carboxylation of structurally distinct VKD proteins associated with a variety of clinical phenotypes, we transiently expressed each of these GGCX mutants into the 3 GGCX-deficient reporter cell lines and determined the efficiency of reporter protein carboxylation. Our results show that most GGCX mutations differentially affected the carboxylation of the reporter proteins (Figure 2B-C; supplemental Table 2). For example, the R83W mutant significantly decreased coagulation factor carboxylation (decreased activity to ∼30%), whereas it only had a minor to moderate effect on the carboxylation of MGP (retained ∼80% activity) and BGP (retained ∼60% activity). Although the R704X mutant had a moderate effect on both coagulation factor and MGP carboxylation (retained ∼50% activity), it abolished BGP carboxylation (<5% activity). Several GGCX mutants (G125R, M174R, D183V, F299S, W315X, Q374X, G537X, and I553fs) abolished carboxylation activity for all 3 reporter proteins, whereas other mutations (D31N, P80L, and W493S) had a negligible effect on all 3 reporter protein carboxylation (supplemental Figure 1). Additionally, the V255M, S284P, R476C/H, W501S, and I532T mutants were fully active or had an increased carboxylation efficiency for some of the reporter proteins. Together, these results indicate that GGCX mutations can differentially affect carboxylation of hepatic and extrahepatic VKD proteins associated with distinct biological functions.
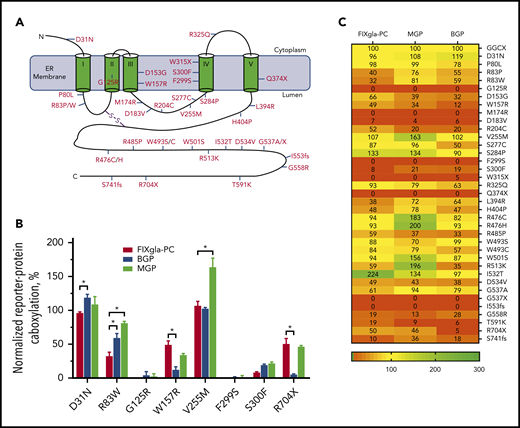
Effect of GGCX mutations on the carboxylation of different VKD reporter proteins. (A) Distribution of naturally occurring mutations in the GGCX molecule. The 5-transmembrane domain topologic structure of GGCX illustrated is based on the model from reference 2 . (B) Carboxylation of the 3 VKD reporter proteins by selected GGCX mutations in HEK293 cells. The GGCX mutation was transiently expressed in GGCX-deficient HEK293 cells stably expressing FIXgla-PC, MGP, or BGP. Transfected cells were incubated with 5 µM vitamin K for 48 hours. Carboxylation efficiency of the reporter protein was determined in the cell culture medium by ELISA. Wild-type GGCX activity was normalized to 100%. Data are presented as the mean ± SD of 3 independent experiments (n = 3). *P < .001. (C) A heat-map representation of the carboxylation activity of the currently identified GGCX mutations for the 3 reporter proteins. Carboxylation activity was determined as described in panel B.
Effect of GGCX mutations on the carboxylation of different VKD reporter proteins. (A) Distribution of naturally occurring mutations in the GGCX molecule. The 5-transmembrane domain topologic structure of GGCX illustrated is based on the model from reference 2 . (B) Carboxylation of the 3 VKD reporter proteins by selected GGCX mutations in HEK293 cells. The GGCX mutation was transiently expressed in GGCX-deficient HEK293 cells stably expressing FIXgla-PC, MGP, or BGP. Transfected cells were incubated with 5 µM vitamin K for 48 hours. Carboxylation efficiency of the reporter protein was determined in the cell culture medium by ELISA. Wild-type GGCX activity was normalized to 100%. Data are presented as the mean ± SD of 3 independent experiments (n = 3). *P < .001. (C) A heat-map representation of the carboxylation activity of the currently identified GGCX mutations for the 3 reporter proteins. Carboxylation activity was determined as described in panel B.
Effect of vitamin K on reporter protein carboxylation by GGCX mutations
Vitamin K administration is the mainstay of therapy in VKCFD.15 However, it is often inefficient in correcting clinical phenotypes associated with defects in MGP and BGP carboxylation.4,25,36 To better understand how clinical phenotypes responded to vitamin K treatment, we determined the EC50 of vitamin K for different reporter protein carboxylation by GGCX mutations. Figure 3A-C shows vitamin K titration examples of wild-type GGCX and of the L394R mutant for carboxylation of the 3 reporter proteins. Although high vitamin K concentrations can partially restore carboxylation of all 3 reporter proteins by the L394R mutant, the EC50 of vitamin K increased 2-, 16-, and 39-fold for the carboxylation of FIXgla-PC, MGP, and BGP, respectively (Table 1). This result suggests that a significantly higher vitamin K concentration is required to rescue the defects of extrahepatic carboxylation of MGP and BGP.
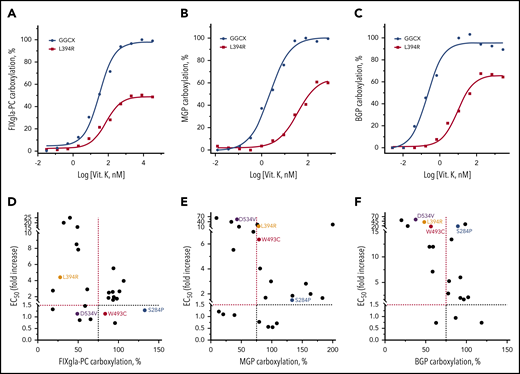
Effect of vitamin K on reporter protein carboxylation by GGCX mutations. (A-C) Vitamin K concentration titration of wild-type GGCX and the L394R mutant for the carboxylation of FIXgla-PC (A), MGP (B), and BGP (C). Wild-type GGCX or the L394R mutant was transiently expressed in GGCX-deficient HEK293 cells stably expressing FIXgla-PC, MGP, or BGP. Reporter-protein carboxylation was determined as described in Figure 1B. (D-F) Correlation between vitamin K’s EC50 and GGCX activity for the carboxylation of FIXgla-PC (D), MGP (E), and BGP (F) by GGCX mutations. The dashed lines indicate the 75% carboxylation activity (x-axis) or a 1.5-fold increase of vitamin K’s EC50. The GGCX mutations discussed in the main text were labeled with different colors.
Effect of vitamin K on reporter protein carboxylation by GGCX mutations. (A-C) Vitamin K concentration titration of wild-type GGCX and the L394R mutant for the carboxylation of FIXgla-PC (A), MGP (B), and BGP (C). Wild-type GGCX or the L394R mutant was transiently expressed in GGCX-deficient HEK293 cells stably expressing FIXgla-PC, MGP, or BGP. Reporter-protein carboxylation was determined as described in Figure 1B. (D-F) Correlation between vitamin K’s EC50 and GGCX activity for the carboxylation of FIXgla-PC (D), MGP (E), and BGP (F) by GGCX mutations. The dashed lines indicate the 75% carboxylation activity (x-axis) or a 1.5-fold increase of vitamin K’s EC50. The GGCX mutations discussed in the main text were labeled with different colors.
Half-maximal stimulation concentrations (EC50, nM) of vitamin K for the carboxylation of 3 different reporter proteins by GGCX mutations
| FIX | MGP | BGP | |
|---|---|---|---|
| GGCX | 4.6 ± 0.3 | 1.9 ± 0.2 | 0.9 ± 0.2 |
| D31N | 3.4 ± 0.4 | 1.4 ± 0.2 | 0.6 ± 0.2 |
| P80L | 8.1 ± 0.9 | 5.7 ± 1.2 | 2.4 ± 0.8 |
| R83P | 115 ± 7 | 42 ± 10 | 10 ± 3 |
| R83W | 93 ± 5 | 7.7 ± 0.8 | 10 ± 4 |
| D153G | 4.1 ± 0.8 | 2.0 ± 0.5 | 312 ± 108 |
| W157R | 73 ± 14 | 75 ± 21 | — |
| R204C | 4.0 ± 0.5 | 2.1 ± 0.8 | 43 ± 17 |
| V255M | 18 ± 2 | 3.8 ± 0.5 | 1.8 ± 0.6 |
| S277C | 3.7 ± 0.6 | 3.1 ± 0.4 | 0.2 ± 0.1 |
| S284P | 5.9 ± 0.8 | 2.9 ± 0.3 | 16 ± 5 |
| S300F | — | 154 ± 58 | >800 |
| R325Q | 7.3 ± 0.8 | 1.5 ± 0.4 | 0.6 ± 0.2 |
| L394R | 10 ± 2 | 30 ± 5 | 35 ± 9 |
| H404P | 39 ± 4 | 236 ± 79 | 443 ± 126 |
| R476C | 26 ± 4 | 3.2 ± 0.5 | 12 ± 3 |
| R476H | 12 ± 1 | 46 ± 5 | 5.2 ± 0.8 |
| R485P | 6.8 ± 0.7 | 11 ± 4 | 294 ± 131 |
| W493C | 5.2 ± 0.4 | 12 ± 3 | 14 ± 5 |
| W493S | 8.5 ± 0.9 | 13 ± 6 | 25 ± 6 |
| W501S | 9.0 ± 1.1 | 5.3 ± 0.9 | 0.8 ± 0.2 |
| R513K | 13 ± 2 | 15 ± 2 | 3.1 ± 1.1 |
| I532T | 13 ± 2 | 3.5 ± 0.5 | 1.4 ± 0.4 |
| D534V | 5.3 ± 1.6 | 104 ± 28 | 46 ± 15 |
| G537A | 13 ± 3 | 1.1 ± 0.4 | 4.5 ± 2.0 |
| G558R | 13 ± 1 | 2.3 ± 1.0 | 17 ± 7 |
| T591K | 6.1 ± 1.5 | 118 ± 39 | — |
| R704X | 36 ± 6 | 22 ± 5 | — |
| S741fs | 4.4 ± 1.4 | 4.0 ± 2.2 | 190 ± 96 |
| FIX | MGP | BGP | |
|---|---|---|---|
| GGCX | 4.6 ± 0.3 | 1.9 ± 0.2 | 0.9 ± 0.2 |
| D31N | 3.4 ± 0.4 | 1.4 ± 0.2 | 0.6 ± 0.2 |
| P80L | 8.1 ± 0.9 | 5.7 ± 1.2 | 2.4 ± 0.8 |
| R83P | 115 ± 7 | 42 ± 10 | 10 ± 3 |
| R83W | 93 ± 5 | 7.7 ± 0.8 | 10 ± 4 |
| D153G | 4.1 ± 0.8 | 2.0 ± 0.5 | 312 ± 108 |
| W157R | 73 ± 14 | 75 ± 21 | — |
| R204C | 4.0 ± 0.5 | 2.1 ± 0.8 | 43 ± 17 |
| V255M | 18 ± 2 | 3.8 ± 0.5 | 1.8 ± 0.6 |
| S277C | 3.7 ± 0.6 | 3.1 ± 0.4 | 0.2 ± 0.1 |
| S284P | 5.9 ± 0.8 | 2.9 ± 0.3 | 16 ± 5 |
| S300F | — | 154 ± 58 | >800 |
| R325Q | 7.3 ± 0.8 | 1.5 ± 0.4 | 0.6 ± 0.2 |
| L394R | 10 ± 2 | 30 ± 5 | 35 ± 9 |
| H404P | 39 ± 4 | 236 ± 79 | 443 ± 126 |
| R476C | 26 ± 4 | 3.2 ± 0.5 | 12 ± 3 |
| R476H | 12 ± 1 | 46 ± 5 | 5.2 ± 0.8 |
| R485P | 6.8 ± 0.7 | 11 ± 4 | 294 ± 131 |
| W493C | 5.2 ± 0.4 | 12 ± 3 | 14 ± 5 |
| W493S | 8.5 ± 0.9 | 13 ± 6 | 25 ± 6 |
| W501S | 9.0 ± 1.1 | 5.3 ± 0.9 | 0.8 ± 0.2 |
| R513K | 13 ± 2 | 15 ± 2 | 3.1 ± 1.1 |
| I532T | 13 ± 2 | 3.5 ± 0.5 | 1.4 ± 0.4 |
| D534V | 5.3 ± 1.6 | 104 ± 28 | 46 ± 15 |
| G537A | 13 ± 3 | 1.1 ± 0.4 | 4.5 ± 2.0 |
| G558R | 13 ± 1 | 2.3 ± 1.0 | 17 ± 7 |
| T591K | 6.1 ± 1.5 | 118 ± 39 | — |
| R704X | 36 ± 6 | 22 ± 5 | — |
| S741fs | 4.4 ± 1.4 | 4.0 ± 2.2 | 190 ± 96 |
—, not detectable; FIX, factor IX.
Next, we correlated the maximum carboxylation activity and the EC50 values of GGCX mutations for the 3 reporter proteins carboxylation. Compared with MGP (Figure 3E) and BGP (Figure 3F) carboxylation, fewer GGCX mutations fell into the top-left corner of the graph for the FIXgla-PC carboxylation (Figure 3D). Additionally, the fold-increase of EC50 (y-axis) for FIXgla-PC carboxylation was significantly smaller than that of MGP and BGP. These results suggest that in FIXgla-PC carboxylation, fewer GGCX mutations have a dramatic effect on both the enzymatic activity and vitamin K’s EC50 than those of MGP and BGP. Additionally, the distributions of individual GGCX mutations on the 3 graphs were significantly different. For example, the W493C mutant only had a minor effect on both the enzymatic activity and vitamin K’s EC50 for FIXgla-PC carboxylation (Figure 3D). However, it significantly increased the EC50 of vitamin K in the carboxylation of MGP (6-fold increase) and BGP (16-fold increase; Figure 3E-F; Table 1). Although the S284P mutant is fully active for carboxylation of all 3 reporter proteins at high vitamin K concentrations, it also has a negligible effect on the EC50 values for FIXgla-PC and MGP carboxylation. It increased the EC50 of vitamin K 18-fold for BGP carboxylation. Overall, these results suggest that naturally occurring GGCX mutations respond differently to vitamin K for the carboxylation of various VKD proteins, which is consistent with clinical observations.17
Characterization of GGCX mutations that dramatically decreased carboxylation of all 3 reporter proteins
Our results show that several GGCX mutants dramatically decreased or abolished the carboxylation of all 3 reporter proteins (Figure 2C; supplemental Table 2). Only a few of these mutants have been characterized. The W315X19 and Q374X36 mutants introduced a stop codon at the mutation sites, which prematurely terminated the GGCX translation at residues W315 and Q374, respectively. Thus, they ablated GGCX activity.47 Characterization of the M174R mutant suggests that it causes GGCX misfolding and degradation.25 To characterize other GGCX mutations that significantly decreased carboxylation of all 3 reporter proteins, we first examined the protein expression levels of these GGCX mutants in HEK293 cells. Results in Figure 4A show that G125R, D183V, G558R, and T591K have eliminated or severely decreased GGCX expressions, which is consistent with results of their significantly decreased carboxylation activity (Figure 2C). This suggests that these mutations probably affect GGCX folding and/or stability.56,57 However, the F299S and S300F mutants, which dramatically decrease reporter protein carboxylation, have a protein expression level similar to that of wild-type GGCX. This suggests that the decreased carboxylation activity of these two mutants might result from the effect of enzyme catalytic activity and/or substrate binding affinity.
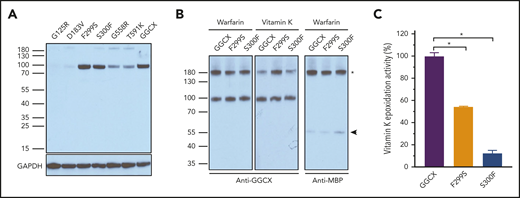
Characterization of GGCX mutations that dramatically decreased carboxylation activity. (A) Western blot analysis of the expression of GGCX mutations in HEK293 cells. Wild-type GGCX and GGCX mutants that significantly decreased reporter protein carboxylation were transiently expressed in GGCX-deficient HEK293 cells for 48 hours. Whole-cell lysate was used for Western blot analysis using a rabbit anti-GGCX polyclonal antibody as the primary antibody. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the sample loading control. (B) Interaction of GGCX with its protein substrate in live cells probed by DSS cross-linking. Wild-type GGCX, the F299S mutant, or the S300F mutant was transiently coexpressed with FIXgla-MBP fusion in GGCX-deficient HEK293 cells. Forty-eight hours later, transfected cells were cross-linked with DSS at a final concentration of 4 mM for 30 minutes. Whole-cell lysate was used for Western blot analysis using a rabbit anti-GGCX or an anti-MBP polyclonal antibody as the primary antibody. The GGCX and FIXgla-MBP cross-linked band was marked with an asterisk. The FIXgla-MBP monomer is marked with an arrowhead. (C) Effect of GGCX mutations on vitamin K epoxidase activity. Wild-type GGCX, the F299S mutant, or the S300F mutant was transiently expressed in GGCX-deficient HEK293 cells. Transfected cells were incubated with vitamin K and the epoxidation of vitamin K to vitamin K epoxide by GGCX was determined by a conventional reversed-phase high-performance liquid chromatography assay. Data are presented as the mean ± SD of 3 independent experiments (n = 3). *P < .001.
Characterization of GGCX mutations that dramatically decreased carboxylation activity. (A) Western blot analysis of the expression of GGCX mutations in HEK293 cells. Wild-type GGCX and GGCX mutants that significantly decreased reporter protein carboxylation were transiently expressed in GGCX-deficient HEK293 cells for 48 hours. Whole-cell lysate was used for Western blot analysis using a rabbit anti-GGCX polyclonal antibody as the primary antibody. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the sample loading control. (B) Interaction of GGCX with its protein substrate in live cells probed by DSS cross-linking. Wild-type GGCX, the F299S mutant, or the S300F mutant was transiently coexpressed with FIXgla-MBP fusion in GGCX-deficient HEK293 cells. Forty-eight hours later, transfected cells were cross-linked with DSS at a final concentration of 4 mM for 30 minutes. Whole-cell lysate was used for Western blot analysis using a rabbit anti-GGCX or an anti-MBP polyclonal antibody as the primary antibody. The GGCX and FIXgla-MBP cross-linked band was marked with an asterisk. The FIXgla-MBP monomer is marked with an arrowhead. (C) Effect of GGCX mutations on vitamin K epoxidase activity. Wild-type GGCX, the F299S mutant, or the S300F mutant was transiently expressed in GGCX-deficient HEK293 cells. Transfected cells were incubated with vitamin K and the epoxidation of vitamin K to vitamin K epoxide by GGCX was determined by a conventional reversed-phase high-performance liquid chromatography assay. Data are presented as the mean ± SD of 3 independent experiments (n = 3). *P < .001.
To further clarify the effect of F299S and S300F on GGCX activity, we used a bifunctional cross-linker, disuccinimidyl suberate (DSS), to monitor the transient enzyme–substrate interaction in live cells by chemical cross-linking. It has been shown that warfarin inhibits the availability of reduced vitamin K (a cofactor for GGCX), thus stabilizing the complex of GGCX and its substrate.58 Vitamin K (required for carboxylation) accelerates the disassociation of the GGCX–substrate complex because of product releasing.59 To explore the effect of F299S and S300F on GGCX’s substrate binding and catalytic activity, we coexpressed these mutants with the coagulation factor in GGCX-deficient HEK293 cells and trapped the enzyme–substrate complex by DSS cross-linking in live cells incubated either with warfarin or vitamin K. Our results show that in the presence of warfarin, F299S and S300F mutants have a similar cross-link efficiency to wild-type GGCX (Figure 4B), suggesting that these 2 mutants have a substrate binding ability similar to the wild-type enzyme. However, in the presence of vitamin K, the cross-linking efficiency significantly decreased for wild-type GGCX and to a lesser extent for the S300F mutant, but not for the F299S mutant (Figure 4B). This result indicates that vitamin K does not accelerate the dissociation of the enzyme–substrate complex of the F299S mutant, suggesting that F299S abolishes carboxylation activity, which agrees with our cell-based activity assay (Figure 2B).
As GGCX is a dual-function enzyme that catalyzes both glutamate carboxylation and vitamin K epoxidation, we explored which activity was affected by the F299S and S300F mutants using our recently established cell-based epoxidation activity assay.45 Results in Figure 4C show that, compared with wild-type GGCX, the S300F mutant decreased epoxidation activity to ∼10%, a level similar to reporter-protein carboxylation in our cell-based study (Figure 2B). This suggests that the S300F mutation mainly affects vitamin K epoxidation activity. However, the F299S mutant, which abolished all reporter protein carboxylation in the cell-based assay (Figure 2B), retained about 50% vitamin K epoxidation activity, suggesting that the F299S mutation affected both glutamate carboxylation and vitamin K epoxidation activities. Together, these results suggest that residues S300 and F299 are essential for vitamin K epoxidation. The location of these 2 residues, within the third transmembrane domain facing the ER lumen,2 is consistent with the hydrophobic characteristics of vitamin K and with the notion that VKD carboxylation occurs in the lumen of the ER.60
GGCX’s C terminus contributes to BGP binding
The R704X mutant (C-terminal truncation from residue 704) abolishes BGP carboxylation while retaining ∼50% activity for FIXgla-PC and MGP carboxylation (Figure 2B), suggesting the important role of GGCX’s C terminus in BGP binding. To test this hypothesis, we used DSS cross-linking to explore the binding of the 3 reporter proteins to the R704X mutant in HEK293 cells. Results in Figure 5A show that MBP fusion reporter proteins of both FIXgla and MGP, but not BGP, can be cross-linked to the R704X mutant, indicating that BGP does not bind to the R704X mutant.
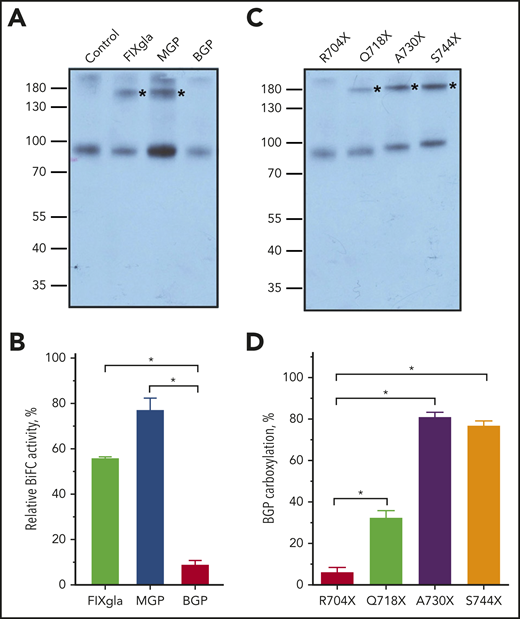
Effect of GGCX’s C terminus on BGP binding. (A) Interaction of the R704X mutant with 3 VKD reporter proteins in live cells probed by DSS cross-linking. The R704X mutant and MBP fusions of the VKD reporter proteins (FIXgla, MGP, BGP) were transiently coexpressed in GGCX-deficient HEK293 cells for DSS cross-linking, as described in Figure 4B. The cross-linked bands of GGCX and its substrate were marked with asterisks. (B) Interaction of the R704X mutant with 3 VKD reporter proteins in live cells probed by the Venus-based BiFC assay. R704X-VN and the reporter protein fused with VC were transiently coexpressed in GGCX-deficient HEK293 cells. Forty-eight hours later, the fluorescence signal was determined directly from the transfected live cells. Data are presented as the mean ± SD of 3 independent experiments (n = 3). *P < .001. (C) Interaction of GGCX C-terminal truncation mutations with BGP probed by DSS cross-linking, as described in panel A. (D) BGP carboxylation by GGCX C-terminal truncation mutations in GGCX-deficient HEK293 cells, as described in Figure 2B. Data are presented as the mean ± SD of 3 independent experiments (n = 3). *P < .001.
Effect of GGCX’s C terminus on BGP binding. (A) Interaction of the R704X mutant with 3 VKD reporter proteins in live cells probed by DSS cross-linking. The R704X mutant and MBP fusions of the VKD reporter proteins (FIXgla, MGP, BGP) were transiently coexpressed in GGCX-deficient HEK293 cells for DSS cross-linking, as described in Figure 4B. The cross-linked bands of GGCX and its substrate were marked with asterisks. (B) Interaction of the R704X mutant with 3 VKD reporter proteins in live cells probed by the Venus-based BiFC assay. R704X-VN and the reporter protein fused with VC were transiently coexpressed in GGCX-deficient HEK293 cells. Forty-eight hours later, the fluorescence signal was determined directly from the transfected live cells. Data are presented as the mean ± SD of 3 independent experiments (n = 3). *P < .001. (C) Interaction of GGCX C-terminal truncation mutations with BGP probed by DSS cross-linking, as described in panel A. (D) BGP carboxylation by GGCX C-terminal truncation mutations in GGCX-deficient HEK293 cells, as described in Figure 2B. Data are presented as the mean ± SD of 3 independent experiments (n = 3). *P < .001.
To further confirm the contribution of GGCX’s C terminus to BGP binding, we used the Venus-based BiFC assay61 to study the interaction between GGCX and its substrate in live cells. Our results, from optimizing the BiFC assay for GGCX and its substrate interactions, show that fusing either the N or C fragment of Venus (VN or VC) to the C terminus of GGCX does not affect GGCX’s activity (supplemental Figure 2A). Additionally, the combination of fusing VN to GGCX and VC to the substrate gives a better signal (supplemental Figure 2B). Using this combination for the BiFC assay, our result shows that the R704X mutant has a significant binding affinity to FIXgla and MGP but not to BGP (Figure 5B). This result agrees with the cell-based activity assay (Figure 2B) and the DSS cross-linking in the enzyme–substrate interaction study (Figure 5A).
Next, we extended the C-terminal truncation of GGCX to residues Q718, A730, or S744 and examined their effect on the binding and carboxylation of BGP. Our results show that a further extension of GGCX C terminus increased BGP binding (Figure 5C) and carboxylation (Figure 5D). However, these C-terminal extension mutants only have minor effects on the carboxylation of either FIXgla or MGP (supplemental Figure 2C-D). Together, these results support the hypothesis that the C terminus of GGCX is essential for BGP binding.
Evaluation of GGCX mutations that affect GGCX’s pre-mRNA splicing
In addition to the missense mutations in GGCX’s exon coding sequences, naturally occurring mutations were also identified in GGCX introns near the intron–exon conjunctions (supplemental Table 3). These mutations were predicted as splice-site mutations that could affect the correct splicing of GGCX’s pre-mRNA. However, only a few of them have been experimentally confirmed. Importantly, previously reported missense mutations in exons near the intron–exon conjunctions could also affect GGCX’s pre-mRNA splicing. To examine the effect of these mutations on GGCX pre-mRNA splicing, we used a minigene splicing assay.47,48
Our results show that previously reported missense mutations G125R (c.373 G>A), G537X (c.1609 G>T), and G537A (c.1610 G>C) induced mis-splicing of GGCX pre-mRNA (Figure 6A). The c.373 G>A mutation results in a splicing product that is ∼200 bp larger than that of wild-type GGCX. The sequencing results show that there is a 211-bp intronic sequence insertion to the spliced mRNA (supplemental Figure 3). However, the c.1610 G>C mutation mainly results in a mRNA product that is ∼150 bp smaller than that of the wild-type GGCX, corresponding to the deletion of exon 12 (supplemental Figure 4). Nevertheless, a small percentage (∼5%) of the correct splicing product was also observed in the c.1610 G>C mutation (Figure 6A). The c.1609 G>T mutation also resulted in 2 splicing products. The major mRNA product is slightly smaller than the wild-type mRNA (Figure 6A), corresponding to the deletion of the 25-bp exonic sequence from exon 11 (Figure 6B). This result is consistent with the result from sequencing the patient’s mRNA.35 The minor mRNA product migrates slightly slower, and its sequencing result shows an insertion of 12 bp from intron 11 (Figure 6C). The corresponding protein sequences of the 2 alternative splicing mRNAs of the c.1609 G>T mutant are shown in Figure 6D.
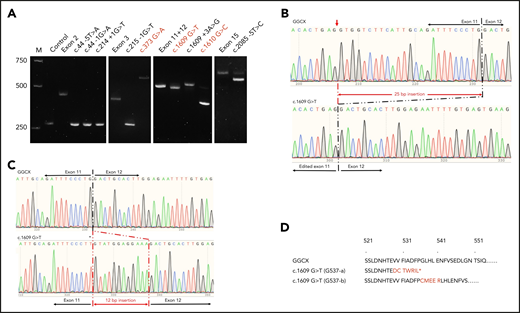
Effect of GGCX mutations on its pre-mRNA splicing. (A) Minigene splicing assay of GGCX mutations. GGCX mutations and the wild-type sequences of the affected exons were transiently transcribed in HEK293 cells. The splicing products were amplified by reverse transcriptase-PCR and visualized by 2.5% agarose gel electrophoresis. Previously reported missense mutations in GGCX’s coding regions were labeled in red. M, DNA ladder marker; Control, splicing product from the empty vector. Exon 2, exon 3, exon 11+12, and exon 15 are the splicing products from the wild-type GGCX sequence of the corresponding exons. (B) Sequencing results of the splicing products of wild-type GGCX and of the c.1609 G>T mutant (bottom band). The mis-splicing site in exon 11 is indicated with a red arrowhead. (C) Sequencing results of the splicing products of wild-type GGCX and the c.1609 G>T mutant (top band). The 12-bp intronic insertion sequences are indicated in red. The c.1609 G>T mutation in the mis-splicing sequence is indicated with an asterisk. (D) Amino acid sequences of the 2 mis-splicing products of the c.1609 G>T mutation. The G537-a sequence corresponds to the mis-splicing of the 25-bp deletion, as shown in panel B. The G537-b sequence corresponds to the mis-splicing of the 12-bp insertion, as shown in panel C. Additional sequences introduced to GGCX are highlighted in red. The asterisk in the G537-a sequence indicates an introduced stop codon because of the frame-shift translation.
Effect of GGCX mutations on its pre-mRNA splicing. (A) Minigene splicing assay of GGCX mutations. GGCX mutations and the wild-type sequences of the affected exons were transiently transcribed in HEK293 cells. The splicing products were amplified by reverse transcriptase-PCR and visualized by 2.5% agarose gel electrophoresis. Previously reported missense mutations in GGCX’s coding regions were labeled in red. M, DNA ladder marker; Control, splicing product from the empty vector. Exon 2, exon 3, exon 11+12, and exon 15 are the splicing products from the wild-type GGCX sequence of the corresponding exons. (B) Sequencing results of the splicing products of wild-type GGCX and of the c.1609 G>T mutant (bottom band). The mis-splicing site in exon 11 is indicated with a red arrowhead. (C) Sequencing results of the splicing products of wild-type GGCX and the c.1609 G>T mutant (top band). The 12-bp intronic insertion sequences are indicated in red. The c.1609 G>T mutation in the mis-splicing sequence is indicated with an asterisk. (D) Amino acid sequences of the 2 mis-splicing products of the c.1609 G>T mutation. The G537-a sequence corresponds to the mis-splicing of the 25-bp deletion, as shown in panel B. The G537-b sequence corresponds to the mis-splicing of the 12-bp insertion, as shown in panel C. Additional sequences introduced to GGCX are highlighted in red. The asterisk in the G537-a sequence indicates an introduced stop codon because of the frame-shift translation.
The intronic mutations of c.44 −5T>A, c.44 −1G>A, c.214 +1G>T, and c.215 −1G>T result in a splicing product similar in size to the control vector (Figure 6A), suggesting that these mutations caused a deletion of the corresponding exons (exon 2 or exon 3) in pre-mRNA splicing (such as exon 2 deletion in supplemental Figure 5). The c.1609 +3A>G mutation results in 2 splicing products, which is similar to the c.1609 G>T mutation. However, the major product is the 12-bp intronic sequence insertion rather than the 25-bp exonic sequence deletion (supplemental Figure 6). The c.2085 −5T>C mutation also results in 2 splicing products (Figure 6A): the major product corresponds to a 41-bp deletion and the minor product corresponds to the correct splicing (supplemental Figure 7).
Discussion
Vitamin K–dependent carboxylation is an essential posttranslational modification required for the biological function of proteins controlling a wide variety of physiologic processes. Carboxylation is accomplished by GGCX, whose naturally occurring mutations result in distinct clinical phenotypes. The goal of this study is to explore the effect of GGCX mutations on the carboxylation of VKD proteins involved in both bleeding and nonbleeding disorders.
Our results show that GGCX mutations differentially affect the carboxylation of various VKD reporter proteins and the efficiency of using vitamin K as a cofactor for their carboxylation (Figures 2 and 3). In general, GGCX mutations have a larger impact on carboxylation of extrahepatic VKD proteins (eg, MGP and BGP) associated with nonbleeding disorders than on those associated with bleeding disorders (eg, coagulation factors). This is consistent with a recent GGCX genotype–phenotype study that suggests patients with skin symptoms often have a less severe bleeding phenotype.17 The significantly increased EC50 values of vitamin K for MGP and BGP carboxylation by some GGCX mutations (Table 1) suggests that higher doses of vitamin K administration are required to recover the extrahepatic carboxylation of MGP and BGP, which is also consistent with clinical observations.17,25 Therefore, results from this study provide clues to the clinical consequences of GGCX mutations and to whether increased vitamin K administration can be useful in ameliorating the pathologic changes of related clinical phenotypes.
For example, the D534V mutation significantly decreased GGCX activity for carboxylation of all 3 VKD reporter proteins (Figure 2C; supplemental Table 2). Although it had only a minor effect on the EC50 of vitamin K for FIXgla-PC carboxylation, the EC50 for MGP and BGP carboxylation increased more than 50-fold (Figure 3D-F; Table 1). These results suggest that the D534V mutation could cause both bleeding and nonbleeding disorders and that vitamin K administration should ameliorate the functional defect of coagulation factors. However, a significantly higher vitamin K concentration is required to rescue the functional defects of MGP and BGP.
The D534V mutation, together with the c.373 G>A (G125R) mutation, was identified as a compound heterozygous mutation in a patient with VKCFD, midfacial hypoplasia, and reduced bone density.19 Our results show that the c.373 G>A mutation is a splice site mutation that prematurely terminates GGCX translation at residue G125 (supplemental Figure 3); this results in an inactive GGCX47 and makes D534V the causative mutation for the patient’s clinical phenotypes. Clinical observations of multiple phenotypes in the patient and that a high dose of vitamin K supplementation rectified the patient’s VKCFD but not the nonbleeding phenotypes19 are consistent with results from our cell-based study.
It has been suggested that some nonbleeding symptoms, such as skin and ocular phenotypes, may be associated with aging rather than with a specific genotype.17 This could be because of an accumulation of certain substances that only lead to symptoms once a critical threshold is reached. Therefore, caution is advised when clarifying clinical observations using results from our cell-based studies. For example, compound heterozygous mutations of R83W and Q374X were found in patients with characteristic features of both PXE and VKCFD.36 As the Q374X mutation abolished GGCX activity,47 R83W is the causative mutation for the clinical phenotypes of the patient. Our cell-based study shows that, compared with wild-type GGCX, the R83W mutant decreased the carboxylation of coagulation factor and MGP to ∼30% and ∼80%, respectively (Figure 2C). Additionally, it increased the EC50 of vitamin K 20-fold for coagulation factor carboxylation but only 4-fold for MGP carboxylation (Table 1). These results suggest that the R83W mutation has a greater impact on carboxylation of coagulation factors than MGP and that MGP carboxylation should be rectified at vitamin K concentrations where the coagulation factor function is corrected.
The patient carrying the R83W and Q374X mutations has a lifelong history of easy bruising, epistaxis, spontaneous gingival bleeding, and prolonged bleeding after dental extractions.62 The patient was initially presented with pubertal menorrhagia at age 13 that required transfusion for anemia; however, no skin manifestations were reported. A daily continuous administration of 5 mg vitamin K ameliorated her bleeding diathesis. However, PXE syndrome was diagnosed at age 27, and progressive facial skin laxity resulted in a face lift procedure at age 39.36 Immunohistochemistry of skin biopsy revealed the deposition of uncarboxylated MGP. These clinical observations are inconsistent with our cell-based study of the R83W mutation, which could be because of the tissue abnormality that developed due to reasons other than defects of VKD carboxylation, such as apoptosis, oxidative stress, or aging.17
Other possible explanations for this discrepancy could be the differences of intestinal absorption and cellular uptake of K vitamins,4,63 mutations in the vitamin K transporter proteins (such as Niemann-Pick C1-Like 1),4 the tissue distribution of different forms of vitamin K, and the different characteristics of hepatic and extrahepatic carboxylation of different VKD proteins. It has been shown that phylloquinone (vitamin K1) is the most abundant form of vitamin K in the liver, whereas menaquinones (vitamin K2) are the main vitamin K in extrahepatic tissues.4,63 Phylloquinone is the primary supplement in the treatment of VKCFD patients for improving coagulation factor carboxylation. However, menaquinones are believed to be important for VKD carboxylation in extrahepatic tissues.64,65 Therefore, caution should be taken when interpreting the coexistence of multiple clinical phenotypes in 1 patient using results from our cell-based studies.
Additionally, several mutations in GGCX’s coding regions near the intron–exon conjunctions were reported as missense mutations (supplemental Table 3). Results from this study show that these mutations affect GGCX pre-mRNA splicing rather than changing the specific residue as missense mutations (Figure 6). Therefore, characterizations of the missense mutations of G125R, G537A, and G537X should not be used for interpreting clinical phenotypes.
In summary, we established cell-based assays to study the clinical consequences of GGCX mutations associated with disorders in blood coagulation, bone formation, and vascular calcification. Systematic characterizations of naturally occurring GGCX mutations with these assays can provide insights into treating patients carrying these mutations. Future studies include the establishment of cell-based assays for other Gla proteins, such as GRP, Gas6, and proline-rich Gla protein that are involved in GGCX associated nonbleeding pathologies (ie, inflammation, cancer cell proliferation, and neurologic disorders).
For publication-related data, please e-mail the corresponding author at jktie@email.unc.edu.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgment
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grant HL131690 (J.-K.T. and D.W.S.).
Authorship
Contribution: Z.H., L.J.S., D.W.S., and J.-K.T. conceived the studies; Z.H. performed all the cell-based functional studies, BiFC assay, chemical cross-linking, and minigene splicing assay; D.-Y.J. created GGCX naturally occurring mutations; X.C. performed epoxidase activity assays; and Z.H. and J.-K.T. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jian-Ke Tie, Department of Biology, University of North Carolina at Chapel Hill, 120 South Rd, Wilson Hall 438, Chapel Hill, NC 27599; e-mail: jktie@email.unc.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal