

Key Points
c-Myc controls HSC self-renewal, quiescence, and survival by regulating Nr4a1, Nr4a2, and Jmjd3 expression.
Apc loss blocks erythroid differentiation by upregulating IL6 secretion in bone marrow endothelial cells, which is mediated by c-Myc.

Abstract
This study was conducted to determine the dosage effect of c-Myc on hematopoiesis and its distinct role in mediating the Wnt/β-catenin pathway in hematopoietic stem cell (HSC) and bone marrow niche cells. c-Myc haploinsufficiency led to ineffective hematopoiesis by inhibiting HSC self-renewal and quiescence and by promoting apoptosis. We have identified Nr4a1, Nr4a2, and Jmjd3, which are critical for the maintenance of HSC functions, as previously unrecognized downstream targets of c-Myc in HSCs. c-Myc directly binds to the promoter regions of Nr4a1, Nr4a2, and Jmjd3 and regulates their expression. Our results revealed that Nr4a1 and Nr4a2 mediates the function of c-Myc in regulating HSC quiescence, whereas all 3 genes contribute to the function of c-Myc in the maintenance of HSC survival. Adenomatous polyposis coli (Apc) is a negative regulator of the Wnt/β-catenin pathway. We have provided the first evidence that Apc haploinsufficiency induces a blockage of erythroid lineage differentiation through promoting secretion of IL6 in bone marrow endothelial cells. We found that c-Myc haploinsufficiency failed to rescue defective function of Apc-deficient HSCs in vivo but it was sufficient to prevent the development of severe anemia in Apc–heterozygous mice and to significantly prolong the survival of those mice. Furthermore, we showed that c-Myc–mediated Apc loss induced IL6 secretion in endothelial cells, and c-Myc haploinsufficiency reversed the negative effect of Apc-deficient endothelial cells on erythroid cell differentiation. Our studies indicate that c-Myc has a context-dependent role in mediating the function of Apc in hematopoiesis.
Introduction
c-Myc, a member of the Myc family of proteins, plays a vital role in many biological activities of cells, such as development, growth, stemness, metabolism, and differentiation.1-4 c-Myc is also a critical gene involved in cell transformation and cancer development,5 including hematological cancers.6-8 c-Myc is required for embryonic hematopoietic development,6,9-11 as well as adult hematopoiesis.12-14 c-Myc has a critical role in regulating the self-renewal and differentiation of HSCs. There is a controversy in literature, however, regarding the effect of c-Myc on these processes. Wilson et al demonstrated that loss of function of c-Myc in mice resulted in the accumulation of HSCs.13 In another study, loss of both c- and N-Myc promoted quick exhaustion of HSCs.12 Forced c-Myc expression in HSCs results in the loss of self-renewal activity.13 It has also been shown that overexpression of c-Myc enhances HSC self-renewal.15 The dosage effect of c-Myc on hematopoiesis remains elusive.
The Wnt/β-catenin pathway is tightly regulated during hematopoiesis, and activation of the pathway disrupts HSC functions.16,17 In our previous study, loss of Apc led to a rapid exhaustion of HSC/hematopoietic progenitor cell (HPC) pools, as well as blockage of multilineage differentiation.18 Apc haploinsufficiency contributes to the development of anemia in mice in a cell-extrinsic manner by affecting the bone marrow (BM) niche,19,20 whereas Apc has an intrinsic role in regulating HSC functions.21,22 However, the role of Apc in the BM niche cells remains unknown.
In this study, we identified an unexpected role of c-Myc haploinsufficiency in regulating HSC self-renewal, quiescence, and survival and identified new downstream mediators of c-Myc in HSCs. More important, we found a new role for Apc in regulating erythropoiesis by controlling secretion of IL6 in endothelial cells. We showed a context-dependent role of c-Myc in mediating the function of Apc in HSCs and endothelial cells. This study provides new insight into the complexity of c-Myc function in hematopoiesis.
Methods
Mice
c-Mycfl/fl mice,1 obtained from The Jackson Laboratory, were crossed with Mx1-Cre mice23 to generate c-Mycfl/+ (WT) and c-Mycfl/+Mx1Cre (HET) mice. c-Myc deletion was induced by injection of 6 mg/kg poly(I:C) IP every other day 3 times. Apcfl/fl mice24 were described in our previous studies. All animal research was approved by the University of Illinois at Chicago and the University of Florida Institutional Animal Care and Use Committees (protocols 16-073 and 201810191, respectively).
Flow cytometric analysis
Suspended single cells were prepared from BM, spleen, thymus, and peripheral blood. Analysis of HSCs, subpopulations of myeloid progenitors, and mature cells were performed as we described previously.18,25 For analysis of cell quiescence (G0 population) and apoptosis, BM cells were stained with Hoechst and pyronin Y or annexin V and 4′6- diamidino-2-phenylindole (DAPI). To perform a BrdU incorporation assay in vivo, BrdU was injected IP for 24 hours before the mice were euthanized. BM cells were collected and stained with antibodies against the cell surface markers (listed in supplemental Table 1, available on the Blood Web site), anti-BrdU antibodies, and DAPI. All cells were analyzed by flow cytometry on a CyAn bench-top analyzer (Beckman Coulter).
BM transplantation
For a competitive repopulation assay, the c-Mycfl/+(WT) and c-Mycfl/+Mx1Cre (HET) mice were euthanized 4 months after polyinosinic-polycytidylic acid (pIpC) induction. BM cells were collected and mixed with an equal number of CD45.1+/CD45.2+ primary BM cells from mice, followed by transplantation into lethally irradiated ly5.1 mice. One month after transplantation, peripheral blood (PB) from each mouse was analyzed by flow cytometry.
In some experiments, BM cells were collected from 6- to 8-week-old nontreated c-Mycfl/+(WT) and c-Mycfl/+Mx1Cre (HET) mice. The deletion of c-Myc was induced 1 month after transplantation.
For the transplantation assay, the BM cells were collected from nontreated 6- to 8-week-old c-Mycfl/+(WT) and c-Mycfl/+Mx1Cre (HET) mice or 6- to 8-week-old ly5.1 mice, followed by transplantation into lethally irradiated ly5.1 mice or c-Mycfl/+(WT) and c-Mycfl/+Mx1Cre (HET) mice. One month after transplantation, pIpC was injected to induce c-Myc deletion.
RNA-sequencing
The LT-HSCs (Lin−Sca1+c-Kit+CD48−CD150+) were sorted from c-Mycfl/+ and c-Mycfl/+Mx1Cre mice (deletion was induced by pIpC) by the MoFlo Astrios Cell Sorter (Beckman Coulter). RNA-sequencing (RNA-seq) was performed at the UCLA Clinical Microarray Core (Los Angeles, CA). Gene Set Enrichment Analysis (GSEA) was performed with GSEA v4.0.0 software, available from the Broad Institute (http://www.broad.mit.edu/gsea/; Massachusetts Institute of Technology, Cambridge, MA).
Real-time PCR
Real-time PCR was performed on a QS3 0.2ML QPCR SYSTEM (Thermo Fisher Scientific). cDNA from HSCs were amplified by the Ovation Pico WTA System V2 (NuGEN Technologies Inc). All primers are listed in supplemental Table 2.
Drug treatment
CX-3543 was purchased from AdooQ BioScience, dissolved in dimethyl sulfoxide (DMSO), and diluted with phosphate-buffered saline. Two months after the induction of c-Myc deletion by pIpC, 12.5 mg/kg CX-3543 was administrated IP 3 times every week until the mice became moribund. DMSO+phosphate-buffered saline served as the negative control.
Immunohistochemistry
Cellular morphology from BM smear, spleen touch, and PB was analyzed by May-Grünwald-Giemsa staining. Spleen sections were stained with hematoxylin and eosin at the core facility at Northwestern University (Chicago, IL). All slides were evaluated in conventional light-field microscopy with an optical microscope (CKX53; Olympus, Tokyo, Japan).
Plasmid resource
The MSCV JMJD3wt plasmid was purchased from Addgene (21212).
Statistical analysis
Statistical significance was calculated with the 2-tailed Student t test. Data are the mean ± standard deviation (SD) of results of triplicate experiments. P < .05 indicated statistically significant results. The Kaplan-Meier survival curve was generated by GraphPad Prism5, and the P value for the survival curve was calculated by the log-rank (Mantel-Cox) test.
BM erythroid progenitor differentiation and culture of endothelial cells in vitro
Endothelial cells were isolated and cultured according to previously published protocols.26,27 When CD31+ cells reached more than 95%, the cells were replated in 24-well plates. One day later, Lin-BM cells isolated from C57B6 WT mice were cultured on endothelial cells in red cell differentiation medium, according to a previously published method.28 Two days after induction, the BM cells were subjected to flow cytometric analysis.
Luciferase assay
Jmjd3, Nr4a1, and Nr4a2 promoter regions were amplified and cloned into the PGL3.0 vector containing a TATA box; primers are listed in supplemental Table 2. The promoter vectors, together with pRL-SV40 which contains Renilla luciferase as the inner control, were transfected into 293T cells, with or without c-Myc overexpression plasmids. Luciferase reporter activity was detected according to the manufacturer’s protocol (Dual-Luciferase Reporter Assay System; Promega).
ChIP assay
The chromatin immunoprecipitation (ChIP) assay was performed according to a previously published method.25 In brief, Lin−BM cells, were harvested and fixed in 1% formaldehyde overnight and then sonicated in radioimmunoprecipitation buffer with a protease inhibitor. A c-Myc antibody (MA1-980; Thermo Fisher Scientific) or control IgG was added with protein A/G and incubated overnight at 4°C. The beads were washed 8 times with radioimmunoprecipitation assay buffer, and DNA was purified with a DNA purification kit (Omega).
Results
Haploinsufficiency of c-Myc leads to ineffective hematopoiesis
To understand the dosage effects of c-Myc in HSCs, we generated a haploinsufficient c-Myc mouse model, c-Mycfl/+ Mx1-Cre, by breeding c-Mycfl/fl mice1 with Mx1-Cre transgenic mice.23 At the age of 6 weeks, deletion of c-Myc was induced by pIpC injection. Consistent with previous studies,14 1 or 2 months after induction of c-Myc deletion, the c-Myc heterozygous mice (c-Myc+/−Mx1-Cre) did not show any abnormal blood counts (supplemental Figure 1A). Interestingly, 4 months after pIpC injection, c-Myc heterozygous mice had significantly decreased white blood cell (WBC) and lymphocyte (LY) counts in PB (Figure 1A), with normal complete blood count results and other parameters, including red blood cells (RBCs) and platelets (data not shown). c-Myc deletion in BM cells was confirmed by PCR (Figure 1B) 4 months after pIpC injection. As determined by flow cytometric analysis, the frequencies of myeloid cells, monocytes, B cells, and red cells in both BM and spleen were comparable (supplemental Figure 1B-I), whereas the frequency of megakaryocytes and macrophages increased in c-Myc heterozygous mice, compared with control mice (supplemental Figure 1J-K). However, total BM cells decreased in c-Myc heterozygous mice (Figure 1C), as well as the total number of myeloid cells, monocytes, and B cells (Figure 1D-F). Interestingly, the frequency of thymus CD4+CD8+ cells decreased (Figure 1G) with comparable thymus weight between WT and c-Myc heterozygous mice (supplemental Figure 1L).

Haploinsufficiency of c-Myc leads to ineffective hematopoiesis. (A) Absolute number of WBCs, neutrophils (NEs), LYs, monocytes (MOs), eosinophils (EOs), and basophils (BAs) in PB from c-Mycfl/+ (WT) and c-Mycfl/+ Mx1-Cre (HET) mice 4 months after pIpC injection (n = 7-8). (B) PCR analysis of the deletion of c-Myc (Mut band) and WT c-Myc allele among genomic DNA in BM cells from WT and HET mice 4 months after pIpC injection. (C) Total BM cells in WT and HET mice 4 months after pIpC injection (n = 6-12). (D-G) Number of myeloid cells (Mac+Gr1+) (D), MOs (Mac+Gr1low) (E), and B cells (B220+) (F) in BM and ratio of CD4+CD8+ cells in the thymus (G) from WT and HET mice 4 months after pIpC injection (n = 7-13). Number of HSCs (I) and HPCs (J) in BM from WT and HET mice 4 months after pIpC injection. Gating strategy is shown in panel H; HSC: Lin−c-Kit+Sca1+ CD48−CD150+; HPC: Lin−c-Kit+Sca1− (n = 10-13). Number of GMPs (L), CMPs (M), and MEPs (N) in BM from WT and HET mice 4 months after pIpC injection. (K) Gating strategy for GMPs: Lin−c-Kit+Sca1− CD16/32+CD34+; CMPs: Lin−c-Kit+Sca1− CD16/32−CD34+; and MEPs: Lin−c-Kit+Sca1− CD16/32−CD34− (n = 8-13). *P < .05; **P < .01; NS nonsignificant, by Student t test.
Haploinsufficiency of c-Myc leads to ineffective hematopoiesis. (A) Absolute number of WBCs, neutrophils (NEs), LYs, monocytes (MOs), eosinophils (EOs), and basophils (BAs) in PB from c-Mycfl/+ (WT) and c-Mycfl/+ Mx1-Cre (HET) mice 4 months after pIpC injection (n = 7-8). (B) PCR analysis of the deletion of c-Myc (Mut band) and WT c-Myc allele among genomic DNA in BM cells from WT and HET mice 4 months after pIpC injection. (C) Total BM cells in WT and HET mice 4 months after pIpC injection (n = 6-12). (D-G) Number of myeloid cells (Mac+Gr1+) (D), MOs (Mac+Gr1low) (E), and B cells (B220+) (F) in BM and ratio of CD4+CD8+ cells in the thymus (G) from WT and HET mice 4 months after pIpC injection (n = 7-13). Number of HSCs (I) and HPCs (J) in BM from WT and HET mice 4 months after pIpC injection. Gating strategy is shown in panel H; HSC: Lin−c-Kit+Sca1+ CD48−CD150+; HPC: Lin−c-Kit+Sca1− (n = 10-13). Number of GMPs (L), CMPs (M), and MEPs (N) in BM from WT and HET mice 4 months after pIpC injection. (K) Gating strategy for GMPs: Lin−c-Kit+Sca1− CD16/32+CD34+; CMPs: Lin−c-Kit+Sca1− CD16/32−CD34+; and MEPs: Lin−c-Kit+Sca1− CD16/32−CD34− (n = 8-13). *P < .05; **P < .01; NS nonsignificant, by Student t test.
We further characterized the HSC/hematopoietic stem progenitors (HSPs) by flow cytometric analysis. Of note, the frequencies of HSCs, hematopoietic progenitor cells (HPCs), common myeloid progenitors (CMPs), and granulocyte-monocyte progenitors (GMPs), but not megakaryocyte-erythroid progenitors (MEPs), decreased slightly (supplemental Figure 1M-O), whereas the total number of HSCs (Figure 1H-I), HPCs (Figure 1H, J), GMPs (Figure 1K-L), and CMPs (Figure 1K, M), but not MEPs (Figure 1K, N), decreased significantly in c-Myc heterozygous mice, but not in control mice. Taken together, our results suggest that c-Myc haploinsufficiency led to a decreased number of HSCs and HPCs, whereas it promoted the differentiation of macrophages and megakaryocytes, but had marginal effects on myeloid, RBC, and B-cell differentiation.
Loss of 1 allele of c-Myc leads to decreased HSC quiescence
Because c-Myc is a critical regulator of the cell cycle and apoptosis,4 we further analyzed the cell cycle dynamics of HSCs and HPCs in c-Myc heterozygous mice. The S phase of LSKs (Lin−c-Kit+Sca+) and LT-HSCs, but not of HPCs, increased significantly in c-Myc heterozygous mice compared with control mice (Figure 2A-B; supplemental Figure 2 A-B). The G0 phase of LSK cells and LT-HSCs decreased markedly, and the G1 phase and G2-S-M phases of HSCs, but not of LSKs, increased in c-Myc heterozygous mice, compared with control mice (Figure 2C-D; supplemental Figure 2 C-D). These results suggest that c-Myc haploinsufficiency promotes the exit of LT-HSCs from quiescence and their entry into cell cycling. Notably, c-Myc heterozygous HSCs and HPCs had an increased frequency of apoptosis compared with control HSCs (Figure 2E-F).
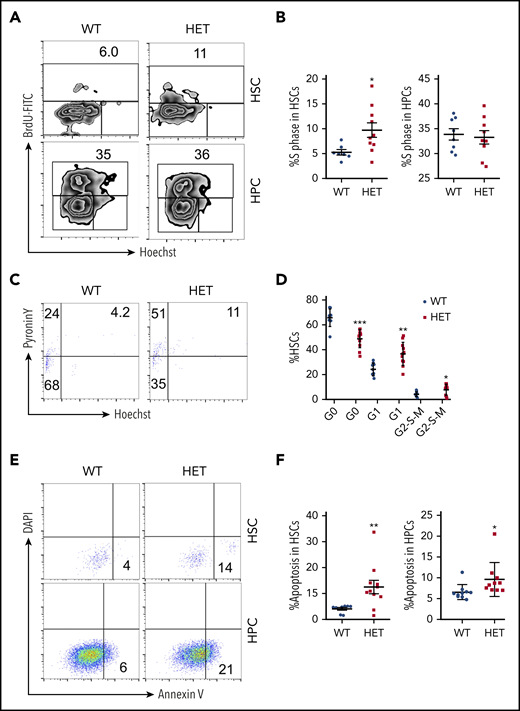
Loss of 1 allele of c-Myc leads to a decrease in HSC quiescence and an increase in HSC apoptosis. (A-B) Flow cytometric analysis of the S phase of HSCs and HPCs from WT and HET mice 4 months after pIpC injection. Cells were stained with BrdU and Hoechst to indicate the S phase (BrdU-FITC+ population). Gating strategy is shown in (A) and summary data of S phase in HSCs and HPCs are presented in (B). (C-D) Flow cytometric analysis of the cell cycle of HSCs from WT and HET mice 4 months after pIpC injection. Cells were stained with Hoechst DNA Dye and pyroninY RNA Dye to indicate the different cell cycle stages. G0 phase (Hoechst− pyroninY−), G1 phase (Hoechst− pyroninY+), and G2-S-M phase (Hoechst+ pyroninY+) cell percentage in the HSC population. Gating strategy (C); summary data (D). (E-F) Flow cytometric analysis of the apoptotic cell ratio (annexin V+) in HSCs and HPCs from WT and HET mice 4 months after pIpC injection (n = 7-14). Gating strategy (E); summary data (F). *P < .05; **P < .01; ***P < .001, by Student t test.
Loss of 1 allele of c-Myc leads to a decrease in HSC quiescence and an increase in HSC apoptosis. (A-B) Flow cytometric analysis of the S phase of HSCs and HPCs from WT and HET mice 4 months after pIpC injection. Cells were stained with BrdU and Hoechst to indicate the S phase (BrdU-FITC+ population). Gating strategy is shown in (A) and summary data of S phase in HSCs and HPCs are presented in (B). (C-D) Flow cytometric analysis of the cell cycle of HSCs from WT and HET mice 4 months after pIpC injection. Cells were stained with Hoechst DNA Dye and pyroninY RNA Dye to indicate the different cell cycle stages. G0 phase (Hoechst− pyroninY−), G1 phase (Hoechst− pyroninY+), and G2-S-M phase (Hoechst+ pyroninY+) cell percentage in the HSC population. Gating strategy (C); summary data (D). (E-F) Flow cytometric analysis of the apoptotic cell ratio (annexin V+) in HSCs and HPCs from WT and HET mice 4 months after pIpC injection (n = 7-14). Gating strategy (E); summary data (F). *P < .05; **P < .01; ***P < .001, by Student t test.
Loss of 1 allele of c-Myc impairs HSC repopulation capacity
A colony-forming assay revealed that c-Myc haploinsufficiency significantly reduced the colony-forming ability of HSPCs (supplemental Figure 3A-B). To investigate the consequence of the loss of a single allele of c-Myc on self-renewal of HSCs in vivo, we performed a competitive repopulation assay, as described in supplemental Figure 4A. The ratio of donor-cell–derived vs competitor-derived cells gradually decreased in c-Myc heterozygous recipient mice, with an average ratio of 1.4:100 6 months after transplantation, whereas the average ratio of donor-cell–derived vs competitor-derived cells (1:1) was unchanged in c-Mycfl/+ recipient mice for up to 6 months after transplantation (Figure 3A-B). The results revealed that all 3 lineages (myeloids, B cells, and T cells) that were derived from c-Myc heterozygous donor cells decreased significantly compared with those that were derived from c-Mycfl/+ donor cells (Figure 3C). Consistent with the observations in PB in recipient mice, the subpopulations of HSPCs, as well as total BM cells that were derived from c-Myc heterozygous donor HSPCs all decreased significantly, compared with those that were derived from c-Mycfl/+ donor cells at 6 months after transplantation (Figure 3D).

Loss of 1 allele of c-Myc impairs HSC repopulation capacity. (A-B) Flow cytometric analysis of donor-derived CD45.2+CD45.1− cells and WT competitor–derived CD45.1+ CD45.2+ cells in recipient WT mice. (A) The ratio of donor-derived to WT competitor–derived cells in PB in recipient mice 1 to 6 months after transplantation; (B) representative plots after transplantation. (C) Flow cytometric analysis of the ratio of donor-derived to WT competitor–derived myeloid cells, B cells, and T cells in PB in recipient mice 6 months after transplantation. (D) Flow cytometric analysis of the ratio of donor-derived to WT competitor-derived cell populations in BM in recipient mice 6 months after transplantation (n = 5). (E-F) Total number of HPCs (E) and HSCs (F) in WT and c-Myc HET–recipient mice at 4 months after induction of c-Myc deletion. (G) Flow cytometric analysis of cell cycle status in WT and c-Myc HET LT-HSCs in recipient mice, as determined by a BrdU incorporation assay (n = 5). (H) Flow cytometric analysis of cell cycle status in WT and c-Myc HET LT-HSCs in recipient mice. Cells were stained with Hoechst DNA dye and pyroninY RNA dye (n = 5). (I) Flow cytometric analysis of the ratio of donor-derived to WT competitor-derived cells in the recipient mice; (J) representative plots are shown 1 and 5 months after transplantation. pIpC was injected 1 month after transplantation. (K) Flow cytometric analysis of the ratio of donor-derived to WT competitor–derived myeloid cells and, B and T cells in PB in the recipient mice 5 months after transplantation (n = 5). *P < .05; **P < .01; ***P < .001, by Student t test.
Loss of 1 allele of c-Myc impairs HSC repopulation capacity. (A-B) Flow cytometric analysis of donor-derived CD45.2+CD45.1− cells and WT competitor–derived CD45.1+ CD45.2+ cells in recipient WT mice. (A) The ratio of donor-derived to WT competitor–derived cells in PB in recipient mice 1 to 6 months after transplantation; (B) representative plots after transplantation. (C) Flow cytometric analysis of the ratio of donor-derived to WT competitor–derived myeloid cells, B cells, and T cells in PB in recipient mice 6 months after transplantation. (D) Flow cytometric analysis of the ratio of donor-derived to WT competitor-derived cell populations in BM in recipient mice 6 months after transplantation (n = 5). (E-F) Total number of HPCs (E) and HSCs (F) in WT and c-Myc HET–recipient mice at 4 months after induction of c-Myc deletion. (G) Flow cytometric analysis of cell cycle status in WT and c-Myc HET LT-HSCs in recipient mice, as determined by a BrdU incorporation assay (n = 5). (H) Flow cytometric analysis of cell cycle status in WT and c-Myc HET LT-HSCs in recipient mice. Cells were stained with Hoechst DNA dye and pyroninY RNA dye (n = 5). (I) Flow cytometric analysis of the ratio of donor-derived to WT competitor-derived cells in the recipient mice; (J) representative plots are shown 1 and 5 months after transplantation. pIpC was injected 1 month after transplantation. (K) Flow cytometric analysis of the ratio of donor-derived to WT competitor–derived myeloid cells and, B and T cells in PB in the recipient mice 5 months after transplantation (n = 5). *P < .05; **P < .01; ***P < .001, by Student t test.
Because homing ability also contributes to HSC engraftment capacity,29 we next evaluated the effect of c-Myc haploinsufficiency on HSPC homing ability. As shown in supplemental Figure 4B-F, there were a comparable frequency and number of WT and c-Myc donor-derived BM and LSK cells in BM from recipient mice 18 hours after transplantation, suggesting that the loss of 1 allele of c-Myc does not affect the homing ability of HSPCs. Together, these results indicate that c-Myc haploinsufficiency significantly disrupts competitive multilineage repopulation capacity of HSCs in vivo.
c-Myc intrinsically regulates HSC quiescence and repopulation capacity
To determine whether c-Myc has an intrinsic role in regulating hematopoiesis and HSC cell cycle in vivo, we generated cohorts of WT recipient mice reconstituted with BM cells from c-Mycfl/+ (WT) and c-Mycfl/+ Mx1Cre (HET) mice (supplemental Figure 5A). As a consequence, recipient mice had a WT HSC microenvironment with c-Myc–haploinsufficient or control HSPCs. Four months after induction of c-Myc deletion, similar to the phenotypes that were observed in primary mice, c-Myc heterozygous recipient mice had reduced BM cellularity (supplemental Figure 5B), with decreased WBC and LY counts (supplemental Figure 5C-D) in PB, as well as a significant decrease in HPCs and HSCs (Figure 3E-F). The c-Myc–haploinsufficient LSKs and HSCs in recipient mice entered cell cycling (Figure 3G-H; supplemental Figure 5E-F).
Next, we investigated whether c-Myc haploinsufficiency intrinsically impairs HSC function in vivo. We generated cohorts of CD45.1 WT recipient mice reconstituted with BM cells from nontreated CD45.2 c-Mycfl/+ (WT) and c-Mycfl/+ Mx1Cre (HET) mice, together with the same number of CD45.2/CD45.1 WT BM cells. One month after transplantation, there were slightly fewer c-Mycfl/+ Mx1Cre donor-cell–derived PB cells compared with control c-Mycfl/+ cell–derived PB cells (Figure 3I; supplemental Figure 5G). At 4 months after induction of c-Myc deletion, the c-Myc–haploinsufficient HSPCs of all 3 lineages showed a significantly decreased capacity for repopulation, with a 50% decrease, compared with control cells (Figure 3I-K).
To determine whether c-Myc has extrinsic effects on hematopoiesis, we generated cohorts of chimeric mice, which had a c-Myc mutant or WT microenvironment with WT BM cells, as described in supplemental Figure 6A. All mice showed normal hematological parameters, with comparable WBC and LY counts (supplemental Figure 6B-C) and cell cycle status of HSCs and LSKs (data not shown), at 4 months after transplantation. As determined by the competitive repopulation assay (supplemental Figure 6A, D), the WT HSPCs, which were exposed to the c-Myc–haploinsufficient or control niche 4 months after transplantation, showed a comparable repopulation capacity in vivo. Taken together, these data indicate that c-Myc intrinsically regulates HSC quiescence and repopulation capacity.
c-Myc haploinsufficiency affects multiple signaling pathways in HSCs
To elucidate the mechanisms of how c-Myc regulates HSC functions, we performed RNA-seq analysis of LT-HSCs, sorted from 3 pairs of c-Mycfl/+ (WT) and c-Mycfl/+Mx1Cre (HET) mice after induction of c-Myc deletion; 196 differentially expressed genes (P < .01; log2-fold change [FC] >0.5) were identified in WT and c-Myc heterozygous HSCs (HET) (Figure 4A; supplemental Table 3). According to gene ontology biological process analysis, the differentially expressed genes are enriched in gene sets involved in regulation of the immune system process, response to stimulus and stress, cell activation, metabolic process, and programmed cell death (Figure 4B). GSEA revealed that the set of genes downregulated in c-Myc heterozygous HSCs showed an enrichment for gene sets that are involved in TNFα and Notch signaling pathways (Figure 4C), but not in the Wnt/β-catenin pathway (data not shown). The TNFα30,31 and Notch32,33 pathways have been implicated in the maintenance of HSCs. Of note, TNF pathway–related genes, including Nfkbid, Tnfaip3, and Tnfaip2, were downregulated significantly in c-Myc–haploinsufficient HSCs (supplemental Figure 7A), suggesting that c-Myc regulates HSC function partially through the TNFα pathway in HSCs. As determined by quantitative RT-PCR (qRT-PCR) assay, Mcl1 was downregulated and caspase 9 was upregulated in c-Myc–haploinsufficient HSCs (supplemental Figure 7A), providing an explanation for the increased apoptosis of c-Myc–haploinsufficient HSCs. Nr4a1/3 deficiency leads to a decrease in HSC quiescence,34 whereas Nr4a2(Nurr1) is highly expressed in quiescent HSCs, and loss of 1 allele of Nurr1 drives HSCs to enter the cell cycle and proliferate.35 As confirmed by qRT-PCR, both Nr4a1 and Nr4a2 were significantly decreased in c-Myc heterozygous HSCs compared with control HSCs (Figure 4D). Jmjd3 is a H3K27me3 demethylase that plays a critical role in regulation of HSC function36 and acute myeloid leukemia leukemogenesis.37 Interestingly, Jmjd3 and its known downstream targets,37 including IL1β and Junb, were all significantly downregulated in c-Myc heterozygous HSCs (Figure 4D). Further study showed that Jmjd3, Nr4a1, and Nr4a2 were also downregulated in multiple potent progenitors, but not in the HPC population (supplemental Figure 7B-C).
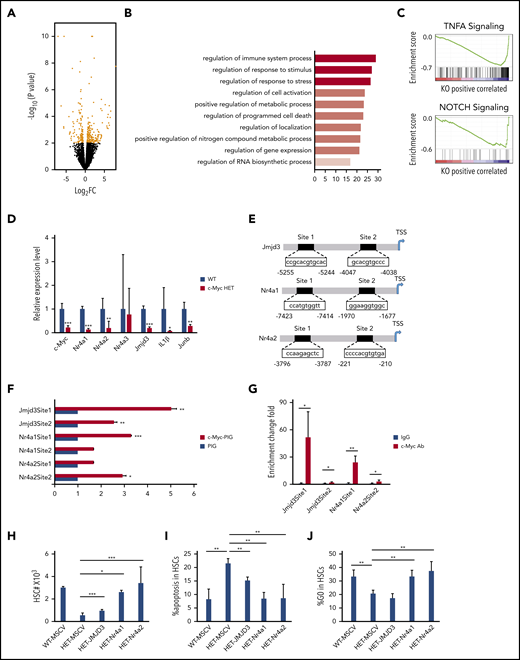
c-Myc regulates multiple pathways related to HSC functions. (A) A volcano plot showing fold changes for differentially expressed genes in LT-HSCs from WT (c-Mycfl/+) and c-Myc HET (c-Mycfl/+Mx1Cre) mice. The experiments were performed in triplicate. (B) Bar graph showing gene ontology biological processes significantly affected in c-Myc HET HSCs. (C) Enrichment plots of selected gene sets from GSEA. Expression data with 20 000 transcripts was used for analysis. (D) qPCR analysis of the expression of selected genes in LT-HSCs. Gene expression was initially normalized to actb expression. Values represent the fold changes in gene expression relative to that in control HSCs. (E) The c-Myc potential binding sites on the promoter region(s) of Jmjd3, Nr4a1, and Nr4a2. (F) Luciferase reporter assays. 293T cells were transfected with Jmjd3, Nr4a1, or Nr4a2 promoter luciferase constructs and control vector PIG or c-Myc–expressing vector. (G) ChIP assay of the binding of endogenous c-Myc to the promoter region(s) of Jmjd3, Nr4a1, and Nr4a2 in Lin− BM cells; IgG served as the negative control. (H-J) Total HSC number (H), the frequency of apoptotic HSCs (I) and the frequency of quiescent HSCs (J), as determined by flow cytometric analysis, in mouse recipients of transplanted WT BM cells expressing MSCV vector, or c-Myc HET BM cells expressing MSCV vector, JMJD3, Nr4a or Nr4a2 (n = 3-6). *P < .05; **P < .01; ***P < .001, by Student t test.
c-Myc regulates multiple pathways related to HSC functions. (A) A volcano plot showing fold changes for differentially expressed genes in LT-HSCs from WT (c-Mycfl/+) and c-Myc HET (c-Mycfl/+Mx1Cre) mice. The experiments were performed in triplicate. (B) Bar graph showing gene ontology biological processes significantly affected in c-Myc HET HSCs. (C) Enrichment plots of selected gene sets from GSEA. Expression data with 20 000 transcripts was used for analysis. (D) qPCR analysis of the expression of selected genes in LT-HSCs. Gene expression was initially normalized to actb expression. Values represent the fold changes in gene expression relative to that in control HSCs. (E) The c-Myc potential binding sites on the promoter region(s) of Jmjd3, Nr4a1, and Nr4a2. (F) Luciferase reporter assays. 293T cells were transfected with Jmjd3, Nr4a1, or Nr4a2 promoter luciferase constructs and control vector PIG or c-Myc–expressing vector. (G) ChIP assay of the binding of endogenous c-Myc to the promoter region(s) of Jmjd3, Nr4a1, and Nr4a2 in Lin− BM cells; IgG served as the negative control. (H-J) Total HSC number (H), the frequency of apoptotic HSCs (I) and the frequency of quiescent HSCs (J), as determined by flow cytometric analysis, in mouse recipients of transplanted WT BM cells expressing MSCV vector, or c-Myc HET BM cells expressing MSCV vector, JMJD3, Nr4a or Nr4a2 (n = 3-6). *P < .05; **P < .01; ***P < .001, by Student t test.
Analysis of public ChIP databases38,39 revealed that there are several c-Myc binding sites on promoter regions of Jmjd3, Nr4a1, and Nr4a2. Two promoter regions of the Jmjd3, Nr4a1, and Nr4a2 genes containing potential binding sites (sites 1 and 2) were subcloned into pGL3.0-TATA which contains a TATA box (Figure 4E). By luciferase reporter and ChIP-qPCR assays, we showed that c-Myc directly bound to the promoter of Jmjd3 (sites 1 and 2), Nr4a1 (site 1), and Nr4a2 (site 2) and activated the expression of these 3 genes (Figure 4F-G). Notably, reexpression of the Nr4a1/Nr4a2 largely reversed the c-Myc haploinsufficiency-induced decrease in HSCs, significantly restored quiescence, and inhibited apoptosis of c-Myc–haploinsufficient HSCs in vivo (Figure 4H-J), whereas reexpression of JMJD3 partially reversed the c-Myc haploinsufficiency-induced decrease in HSCs and inhibited apoptosis, but did not restore the quiescence of c-Myc–haploinsufficient HSCs (Figure 4H-J). These results indicate that Nr4a1 and Nr4a2 are critical downstream mediators of c-Myc in regulating HSC quiescence, whereas all 3 genes, Nr4a1, Nr4a2, and Jmjd3, partially contribute to the role of c-Myc in the regulation of HSC survival. The evidence, taken together, shows that c-Myc haploinsufficiency affects the functions of HSCs through multiple downstream pathways.
c-Myc haploinsufficiency rescues Apc-heterozygous deletion-induced anemia
c-Myc has been reported to be a key downstream target of the Wnt/β-catenin pathway.5,40 To determine whether c-Myc mediates the Wnt/β-catenin signaling pathway in the hematopoietic system as well, we generated a cohort of Apcfl/+c-Mycfl/+ (WT), Apcfl/+Mx1Cre (A), Apcfl/+c-Mycfl/+ Mx1Cre (AC), and c-Mycfl/+ Mx1Cre (C) mice by crossing Apcfl/+ with c-Mycfl/+ Mx1Cre mice . Deletion of Apc and c-Myc was induced by pIpC injection, and deletion efficiency was confirmed by PCR analysis. Consistent with our previously published data,21 Apc mutant mice developed severe anemia, and all mice died within 8 months (Figure 5A). Interestingly, none of the Apc and c-Myc double-heterozygous mice died within 8 months, and the mice showed normal complete blood count parameters (Figure 5B-E; supplemental Figure 8A-K). Thus, c-Myc haploinsufficiency prevented Apc-heterozygous loss–induced macrocytic anemia. Loss of a single allele of Apc resulted in the blockage of erythroid cell differentiation at the proerythroblast (R1, Ter119medCD71hi) and basophilic erythroblast (R2, Ter119hiCD71hi) early stages,21 which was reversed by c-Myc heterozygous deletion (Figure 5F-G; supplemental Figure 8L-M). In addition, whereas the mutant Apc mice developed splenomegaly with an absence of white pulp and an expansion of red pulp (Figure 5H-I), Apc+/−c-Myc+/− mutant mice showed normal size and structure of the spleen, compared with WT control mice (Figure 5 H-I), including the normal peripheral blood smear (Figure 5I). Together, our data suggest that c-Myc haploinsufficiency reversed the blockage of erythroblast cell differentiation induced by heterozygous loss of Apc.
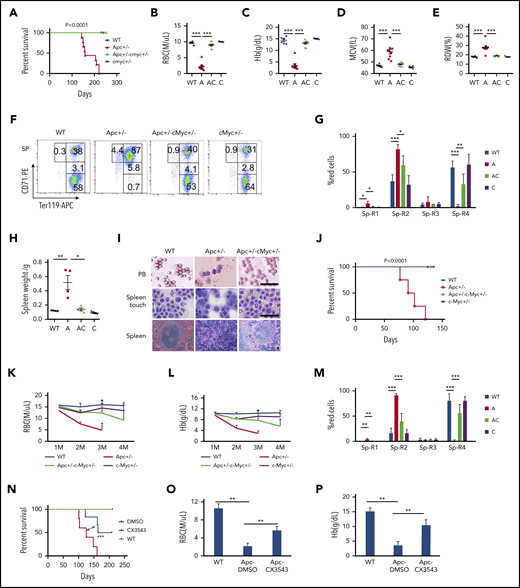
c-Myc haploinsufficiency rescues Apc-heterozygous deletion-induced anemia. (A) Kaplan-Meier survival curve of WT (Apcfl/+c-Mycfl/+), Apc+/− (A, Apcfl/+ Mx1Cre), Apc+/−c-Myc+/− (AC, Apcfl/+c-Mycfl/+ Mx1Cre), and c-Myc+/−(C, c-Mycfl/+ Mx1Cre) mice (n = 6-9). The graph starts from the first day after the third pIpC injection. (B-E) Absolute number of RBCs (B), Hb level (C), mean corpuscular volume (MCV) (D), and RBC distribution width (RDW) (E) in PB in each group of mice (n = 6-9). Data were collected when the first Apc+/− mice become moribund. (F-G) Flow cytometric analysis of erythroid (Ter119+) in spleen (Sp) from WT (Apcfl/+c-Mycfl/+), Apc+/−, Apc+/−c-Myc+/−, and c-Myc+/− mice. Gating strategy (F) and summary data (G) (n = 4). (H) Spleen weights for each cohort (n = 4). (I) Histological analysis of PB smear, spleen touch, and spleen section from WT (Apcfl/+c-Mycfl/+), Apc+/−, and Apc+/−c-Myc+/− mice. PB and spleen touch, May-Grünwald-Giemsa stain; spleen, hematoxylin and eosin. Scale bars, 50 μm. (J) Kaplan-Meier survival curve of WT control, Apc+/−, Apc+/−c-Myc+/−, and c-Myc +/− mice reconstituted with WT CD45.1 BM cells (n = 4-7). The graph starts from the first day after the third pIpC injection. The number of RBCs (K) and Hb level (L) in each cohort of mice from the first to the fourth month after transplantation (n = 4-7). (M) Flow cytometric analysis of erythroid blast cells in spleen from each cohort of mice (n = 4). (N) Kaplan-Meier survival curve of WT and Apc+/− mice reconstituted with WT CD45.1 BM cells after receiving 10 Gy irradiation. WT mice were treated with vehicle control and Apc+/− mice were treated with vehicle control or CX-3543 (n = 5-6). CX-3543 was administered for 3 times every week after 2 months of pIpC-induced deletion until mice were euthanized. The graph starts from the first day after the third pIpC injection. Absolute number of RBCs (O) and the Hb level (P) in WT mice treated with vehicle control or Apc+/− mice treated with vehicle control or CX-3543 (n = 5-6). Data were measured at the time when the first Apc+/− mouse become moribund. *P < .05; **P < .01; ***P < .001, by Student t test or log-rank (Mantel-Cox) test for survival curve.
c-Myc haploinsufficiency rescues Apc-heterozygous deletion-induced anemia. (A) Kaplan-Meier survival curve of WT (Apcfl/+c-Mycfl/+), Apc+/− (A, Apcfl/+ Mx1Cre), Apc+/−c-Myc+/− (AC, Apcfl/+c-Mycfl/+ Mx1Cre), and c-Myc+/−(C, c-Mycfl/+ Mx1Cre) mice (n = 6-9). The graph starts from the first day after the third pIpC injection. (B-E) Absolute number of RBCs (B), Hb level (C), mean corpuscular volume (MCV) (D), and RBC distribution width (RDW) (E) in PB in each group of mice (n = 6-9). Data were collected when the first Apc+/− mice become moribund. (F-G) Flow cytometric analysis of erythroid (Ter119+) in spleen (Sp) from WT (Apcfl/+c-Mycfl/+), Apc+/−, Apc+/−c-Myc+/−, and c-Myc+/− mice. Gating strategy (F) and summary data (G) (n = 4). (H) Spleen weights for each cohort (n = 4). (I) Histological analysis of PB smear, spleen touch, and spleen section from WT (Apcfl/+c-Mycfl/+), Apc+/−, and Apc+/−c-Myc+/− mice. PB and spleen touch, May-Grünwald-Giemsa stain; spleen, hematoxylin and eosin. Scale bars, 50 μm. (J) Kaplan-Meier survival curve of WT control, Apc+/−, Apc+/−c-Myc+/−, and c-Myc +/− mice reconstituted with WT CD45.1 BM cells (n = 4-7). The graph starts from the first day after the third pIpC injection. The number of RBCs (K) and Hb level (L) in each cohort of mice from the first to the fourth month after transplantation (n = 4-7). (M) Flow cytometric analysis of erythroid blast cells in spleen from each cohort of mice (n = 4). (N) Kaplan-Meier survival curve of WT and Apc+/− mice reconstituted with WT CD45.1 BM cells after receiving 10 Gy irradiation. WT mice were treated with vehicle control and Apc+/− mice were treated with vehicle control or CX-3543 (n = 5-6). CX-3543 was administered for 3 times every week after 2 months of pIpC-induced deletion until mice were euthanized. The graph starts from the first day after the third pIpC injection. Absolute number of RBCs (O) and the Hb level (P) in WT mice treated with vehicle control or Apc+/− mice treated with vehicle control or CX-3543 (n = 5-6). Data were measured at the time when the first Apc+/− mouse become moribund. *P < .05; **P < .01; ***P < .001, by Student t test or log-rank (Mantel-Cox) test for survival curve.
Reduced expression of c-Myc in the BM niche prevents Apc haploinsufficiency-induced severe anemia in mice
Because Apc loss-induced anemia was caused by a cell-extrinsic mechanism,20,41 we sought to determine whether c-Myc heterozygous deletion rescued Apc loss-induced anemia by a niche-mediated mechanism. After induction of deletion of c-Myc and Apc by pIpC induction, Apcfl/+c-Mycfl/+ (WT), Apcfl/+Mx1Cre (A), Apcfl/+c-Mycfl/+ Mx1Cre (AC), and c-Mycfl/+ Mx1Cre (C) mice were subjected to 10 Gy irradiation, followed by transplantation with CD45.1 WT BM cells (supplemental Figure 9A). The resultant chimeric WT control, Apc+/−, Apc+/−c-Myc+/−, and c-Myc +/− mice had a WT or mutant microenvironment with WT BM cells. Consistent with previous reports,19,42 Apc+/− chimeric mice with Apc mutant niche cells developed anemia, and all mice died within 4 months after transplantation (Figure 5J). However, all Apc+/−c-Myc+/− and c-Myc +/− mice survived at this time point without development of anemia (Figure 5J), as evidenced by the presence of normal RBC and hemoglobin (Hb) level in PB (Figure 5K-L). Flow cytometric analysis of erythroblast cells in BM and SP cells revealed that c-Myc haploinsufficiency in niche cells reversed Apc-loss–induced blockage of erythroblast cell differentiation, whereas loss of a single allele of c-Myc in niche cells did not affect erythroblast cell differentiation in BM and SP (Figure 5M; supplemental Figure 9B). Double-mutant mice showed a normal spleen weight and size (supplemental Figure 9C-D), as well as normal BM suspension (supplemental Figure 9E). These data suggest that c-Myc haploinsufficiency rescues Apc loss-induced anemia through a niche-mediated mechanism.
To assess whether inhibition of c-Myc by a pharmacological inhibitor reverses Apc-loss–induced anemia in mice, we treated Apc-haploinsufficient mice 3 times a week with CX-3543 (Quarfloxin), which is a small-molecule compound that targets the Myc G-quadruplex site.43-46 As shown in supplemental Figure 10A-B, CX-3543 treatment significantly induced apoptosis in primary LSKs whereas c-Myc overexpression fully rescued CX-3543-induced apoptosis, suggesting that the inhibitory effect of CX-3543 is mainly mediated by c-Myc. Notably, CX-3543 treatment significantly prolonged the survival of Apc-haploinsufficient mice (Figure 5N). After 2 months of administration, Apc-haploinsufficient mice displayed significantly improved RBC (Figure 5O) and Hb (Figure 5P) parameters, compared with nontreated mice, indicating that pharmacological inhibition of c-Myc delayed the development of anemia induced by Apc loss in mice.
Loss of Apc induces IL6 secretion in BM endothelial cells, leading to a blockage of erythroid differentiation
The BM niche has complex components. Single-cell analysis shows that there are nearly 17 different cell populations in the BM niche.47 Expression of Cre is induced in mesenchymal stem cells (MSCs) and endothelial cells in Mx1-Cre transgenic mice.48 MSCs and endothelial cells were isolated from WT and Apc-haploinsufficient mice and cultured in vitro, as previously described.26,27,49,50 As determined by qPCR analysis, Apc haploinsufficiency resulted in upregulation of c-Myc in endothelial cells but not in MSCs (Figure 6A-B). We next cocultured endothelial cells isolated from WT and Apc-haploinsufficient mice with primary WT BM progenitor cells and induced progenitor cells to differentiate into erythroid cells in vitro according to a previously established protocol.28 Compared with WT endothelial cells, Apc-haploinsufficient endothelial cells significantly delayed progenitor cell differentiation, as evidenced by the higher frequency of the early stage of erythroblasts (Ter119+CD71+) and a lower frequency of late stages of erythroblasts (Ter119+CD71−) with fewer visible RBCs in BM-derived progenitors cocultured with Apc-haploinsufficient endothelial cells, compared with WT endothelial cells (Figure 6C-E). To investigate how endothelial cells affect erythroid cell differentiation, a panel of cytokines was screened in the endothelial cell culture medium. Surprisingly, IL6 was significantly increased by 10-fold on average in Apc+/− endothelial cell medium (Figure 6F). We showed that IL6 inhibited erythroid cell differentiation in a dose-dependent manner in vitro (Figure 6G). To determine whether c-Myc loss can reverse erythroid differentiation blockage induced by Apc-haploinsufficient endothelial cells, the endothelial cells were isolated from WT, Apc+/−, Apc+/−c-Myc+/−, and c-Myc+/− mice and cocultured with primary BM progenitor cells. We found that deletion of 1 allele of c-Myc prevented upregulation of IL6 secretion in Apc-haploinsufficient endothelial cells and fully rescued erythroid differentiation blockage induced by Apc-haploinsufficient endothelial cells (Figure 6H-I). These data suggest that endothelial cells are involved in the regulation of erythroid cell differentiation and that Apc loss-induced blockage of erythroid cell differentiation is at least partially mediated by upregulation of IL6 as a consequence of Apc loss-induced c-Myc expression in endothelial cells.
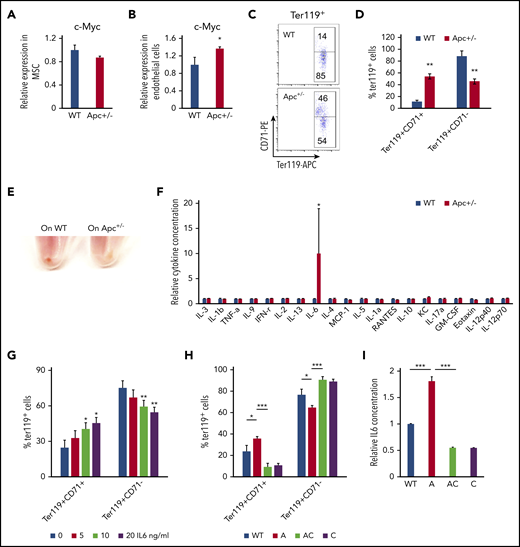
Endothelial cells in Apc-haploinsufficient mice block erythroid differentiation through IL6 secretion. qPCR analysis of c-Myc expression in MSCs (A) and endothelial cells (B) isolated from WT and Apc+/− mice. (C-D) Flow cytometric analysis of erythroid cells in Lin− BM cells cocultured with the endothelial cells from WT or Apc+/− mice in erythroid differentiation medium. Gating strategy (C); summary data (D). (E) Cell pellets of Lin− BM cells cocultured with the endothelial cells from WT or Apc+/− mice in erythroid differentiation medium. (F) Detection of cytokines in the supernatant of WT and Apc+/− endothelial cells. (G) Flow cytometric analysis of erythroid cells in Lin− BM treated with different concentrations of IL6. (H) Flow cytometric analysis of erythroid cells in Lin− BM cocultured with endothelial cells from WT, Apc+/−, Apc+/−cMyc+/−, or c-Myc+/− mice in erythroid differentiation medium. (I) Detection of IL6 concentration in the supernatant of WT, Apc+/−, Apc+/−cMyc+/−, or c-Myc+/− endothelial cells. *P < .05; **P < .01; ***P < .001, by Student t test.
Endothelial cells in Apc-haploinsufficient mice block erythroid differentiation through IL6 secretion. qPCR analysis of c-Myc expression in MSCs (A) and endothelial cells (B) isolated from WT and Apc+/− mice. (C-D) Flow cytometric analysis of erythroid cells in Lin− BM cells cocultured with the endothelial cells from WT or Apc+/− mice in erythroid differentiation medium. Gating strategy (C); summary data (D). (E) Cell pellets of Lin− BM cells cocultured with the endothelial cells from WT or Apc+/− mice in erythroid differentiation medium. (F) Detection of cytokines in the supernatant of WT and Apc+/− endothelial cells. (G) Flow cytometric analysis of erythroid cells in Lin− BM treated with different concentrations of IL6. (H) Flow cytometric analysis of erythroid cells in Lin− BM cocultured with endothelial cells from WT, Apc+/−, Apc+/−cMyc+/−, or c-Myc+/− mice in erythroid differentiation medium. (I) Detection of IL6 concentration in the supernatant of WT, Apc+/−, Apc+/−cMyc+/−, or c-Myc+/− endothelial cells. *P < .05; **P < .01; ***P < .001, by Student t test.
Reduced c-Myc expression failed to rescue the self-renewal defect of Apc-haploinsufficient HSCs in vivo
We next determined whether c-Myc mediates the role of Apc in HSCs in a cell-intrinsic manner. We performed a competitive repopulation assay, using BM cells isolated from pIpC-treated Apcfl/+c-Mycfl/+ (WT), Apcfl/+Mx1Cre (A), and Apcfl/+c-Mycfl/+ Mx1Cre (AC) mice, as described in supplemental Figure 11. Consistent with our previously published results,21 Apc+/− HSPCs had a gradually reduced cell repopulation capacity compared with WT donor-derived cells (Figure 7A-B). Notably, Apc and c-Myc double-heterozygous HSPCs had less repopulation of all 3 lineages than Apc-heterozygous HSPCs did in vivo in recipient mice (Figure 7A-D). Together, our data show that loss of a single allele of c-Myc could not rescue the repopulation defect of Apc+/− HSPCs. Instead, heterozygous loss of both Apc and c-Myc had an additively negative effect on self-renewal capacity of HSPCs.
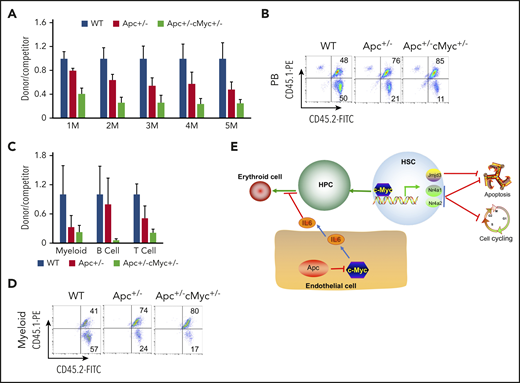
Reduced expression of c-Myc does not rescue the self-renewal defect of HSCs in mice. (A) Flow cytometric analysis of donor (CD45.2+CD45.1−)– and WT competitor (CD45.2+CD45.1+)–derived PB cells in recipient mice (CD45.2−CD45.1+) 1 to 5 months after transplantation. (B) Representative diagrams of PB flow cytometry data for the fifth month. (C) Flow cytometric analysis of donor– and WT competitor–derived PB cells in different lineage populations from the recipient mice. (D) Representative diagram of flow cytometric analysis of myeloid cells in mice 5 months after transplantation (n = 5 for each group). (E) A model of c-Myc–mediated intrinsic and extrinsic regulation of HSC fate. c-Myc mediates the function of the Wnt/β-catenin signaling pathway in endothelial cells through IL6 secretion. c-Myc controls HSC quiescence and quiescence by directly activating expression of Nr4a1, Nr4a2, and Jmjd3.
Reduced expression of c-Myc does not rescue the self-renewal defect of HSCs in mice. (A) Flow cytometric analysis of donor (CD45.2+CD45.1−)– and WT competitor (CD45.2+CD45.1+)–derived PB cells in recipient mice (CD45.2−CD45.1+) 1 to 5 months after transplantation. (B) Representative diagrams of PB flow cytometry data for the fifth month. (C) Flow cytometric analysis of donor– and WT competitor–derived PB cells in different lineage populations from the recipient mice. (D) Representative diagram of flow cytometric analysis of myeloid cells in mice 5 months after transplantation (n = 5 for each group). (E) A model of c-Myc–mediated intrinsic and extrinsic regulation of HSC fate. c-Myc mediates the function of the Wnt/β-catenin signaling pathway in endothelial cells through IL6 secretion. c-Myc controls HSC quiescence and quiescence by directly activating expression of Nr4a1, Nr4a2, and Jmjd3.
Discussion
Before our study, the dosage effect of c-Myc in adult stem cells was poorly understood. In this study, we showed a significant dosage effect of c-Myc on the maintenance of HSCs, and elucidated previously unrecognized downstream pathways that mediate the role of c-Myc in HSCs.
In contrast to a previous report that depletion of c-Myc led to an accumulation of HSCs13 as a consequence of blockage of HSC differentiation, we found that haploinsufficiency of c-Myc reduced quiescence and induced apoptosis of HSCs, leading to a decrease in HSCs and impaired HSC self-renewal capacity, but did not block HSPC differentiation. In contrast, we showed that c-Myc haploinsufficiency increased the frequency of megakaryocytes, which is consistent with a previous study.14 In addition, we showed that c-Myc haploinsufficiency increased the frequency of macrophages, suggesting a possible role of c-Myc in regulating macrophage differentiation.
Previous studies showed that impaired differentiation of c-Myc–deficient HSCs is related to the prevention of HSC release from niche cells,13 which is associated with upregulation of N-cadherin and integrins in c-Myc–deficient HSCs.13 However, we found that expression of N-cadherin and integrins was comparable in c-Myc heterozygous and control HSCs by RNA-seq analysis, supporting our observation that the defective function of c-Myc–haploinsufficient HSCs is intrinsic. At the molecular level, we showed that Nr4a1, Nr4a2, and Jmjdc3, which are important regulators of HSC functions,34,35,37 are the previously unnoticed targets of c-Myc in HSCs and that they mediate the in vivo function of c-Myc in regulating HSC quiescence, survival, and self-renewal (Figure 7E). TNFα and Notch pathways, which are critical for the maintenance of HSCs,30-33 were also deregulated in c-Myc–haploinsufficient HSCs. However, their role in mediating the function of c-Myc in HSPCs remains to be determined by additional experiments in the future. Previous studies showed that c- and N-mycs are expressed in complementary patterns during embryonic development.51,52 However, no significant compensatory upregulation of c- or N-myc transcripts was detected in the N-myc– or c-Myc–deficient LSK population,12,13 respectively. In consistence, as determined by RNA-seq analysis, N-myc was not significantly upregulated in c-Myc–haploinsufficient HSCs compared with WT HSCs (data not shown).
Previous studies have shown that Apc intrinsically regulates HSC functions, whereas it controls erythroid cell differentiation through its function in BM niche cells.19,21,41 It has been shown that activation of the Wnt/β-catenin signaling in osteoblasts induces the malignant changes in HSPCs.53 However, the mechanism of how the Wnt/β-catenin signaling pathway contributes to the regulation of erythropoiesis remains elusive. We have provided the first evidence that heterozygous deletion of Apc significantly increases secretion of inflammatory cytokine IL6 in BM endothelial cells. IL6 is negatively associated with anemia in many diseases54-56 and significantly inhibits human TF-1 cell line erythroid maturation.57 A recent study showed that human acute myeloid leukemia cell–secreted IL6 blocks erythroid differentiation in vitro and in vivo.58 Consistently, we found that Apc-haploinsufficient endothelial cells suppressed erythroid differentiation through upregulation of IL6 secretion. c-Myc is a key downstream target gene in the Wnt pathway.5,40 We found that loss of 1 allele of c-Myc rescued Apc haploinsufficiency–induced anemia. Our results showed that Apc loss upregulated c-Myc expression, which mediated Apc haploinsufficiency–induced IL6 secretion in BM endothelial cells and contributed to the blockage of erythroid progenitor cell differentiation by Apc-haploinsufficient endothelial cells, providing a new mechanistic insight into the role of c-Myc in Apc haploinsufficiency–induced infective erythropoiesis (Figure 7E). Notably, c-Myc haploinsufficiency failed to rescue the functional defects of Apc-heterozygous HSCs. Thus, our findings suggest that c-Myc mediates the role of the Wnt/β-catenin pathway in hematopoiesis in a cell-context–dependent manner.
The RNA-seq data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE156361). Other data are available in the NCBI BioProject database (accession number PRJNA657604).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by National Institutes of Health (NIH), National Heart, Lung, and Blood Institute grant R01HL131444 (Z.Q.) and National Institute of Diabetes and Digestive and Kidney Diseases grant R01DK107615 (Z.Q.), and by a Leukemia Lymphoma Society scholar award (Z.Q.).
Authorship
Contribution: Z.Q. designed the experiments; Y.S., R.M., Q.W., S.Z., and C.Y. performed the experiments; Z.Q. and Y.S. analyzed the data; Z.Q. and Y.S. wrote the manuscript; K.P., J.Z., H.N., Y.H., and Y.Z. provided advice and new reagents and analytic tools; and all authors critically reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Zhijian Qian, Department of Medicine, UF Health Cancer Center, University of Florida, 2033 Mowry Rd, Rm 257, Gainesville, FL 32610; e-mail: zhijian.qian@medicine.ufl.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal