

In this issue of Blood, 1 reveal that in sickle cell disease (SCD), hemolysis-derived heme triggers release of IFN-I, which ultimately results in increased liver macrophages that phagocytize antibody-coated erythrocytes (see figure) in both in vivo (mouse) and in vitro (human) models.
Heme is a crucial cofactor in aerobic metabolism. It fulfills its functions when bound to proteins and compartmentalized within cells. In vertebrates, heme is mostly found in erythrocytes where it performs gas transport as a part of hemoglobin. Despite its essential role, heme has a dark side. The presence of redox-active iron ion and its extensive hydrophobicity make it an inherently dangerous and toxic molecule.2 Thus, heme manifests strong prooxidative and proinflammatory properties; it can interfere with coagulation and complement cascades and oxidize plasma lipoproteins in blood.3,4 These activities of heme are particularly important when it is not bound to a protein. Considerable quantities of heme can be released as a result of hemolysis in various pathological conditions, such as SCD, thalassemia, hemolytic uremic syndrome, and autoimmune hemolytic anemia, among others. The capacity of cell-free heme to trigger proinflammatory states in different cell types, such as neutrophils, macrophages, endothelial cells, and platelets, and to activate the complement system has been postulated to contribute directly to the pathogenesis of the hemolytic diseases.3,4 Mechanistically, it has been shown that heme serves as a ligand for the prototypic proinflammatory receptor TLR45 and has the capacity to activate the inflammasome.6 Extracellular heme can also trigger indirect proinflammatory effects. For example, by activating the complement system, heme can induce release in plasma of C3a and C5a, which have potent proinflammatory actions.4,6
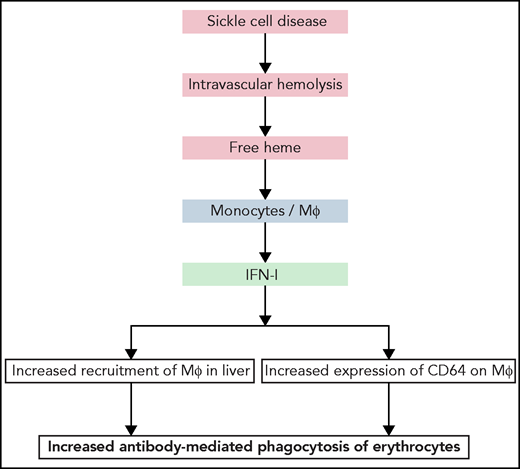
Summary of the principal findings of the work of Liu et al. Hemolytic disorders, such as SCD, are characterized by intravascular hemolysis, leading to release of cell-free heme. Liu et al show that free heme can act on monocyte and macrophages in liver, triggering release of IFN-I. The latter can induce further recruitment of monocytes and their differentiation in the liver into macrophages proficient at phagocytosis of erythrocytes. Moreover, IFN-I induced expression of IgG Fc-binding receptor CD64 on macrophages. Both the increased number of macrophages in liver and higher expression of high-affinity IgG receptor can boost phagocytosis and destruction of erythrocytes.
Summary of the principal findings of the work of Liu et al. Hemolytic disorders, such as SCD, are characterized by intravascular hemolysis, leading to release of cell-free heme. Liu et al show that free heme can act on monocyte and macrophages in liver, triggering release of IFN-I. The latter can induce further recruitment of monocytes and their differentiation in the liver into macrophages proficient at phagocytosis of erythrocytes. Moreover, IFN-I induced expression of IgG Fc-binding receptor CD64 on macrophages. Both the increased number of macrophages in liver and higher expression of high-affinity IgG receptor can boost phagocytosis and destruction of erythrocytes.
In this study, Liu et al discovered a novel effect of heme on the immune system: it triggers expression of IFN-I. They confirm that in patients with SCD and in a mouse model of SCD, significantly higher levels of IFN-α are present compared with healthy individuals or wild-type (WT) mice.7 Importantly, they found a strong correlation between the levels of extracellular heme and concentration of IFN-α in the plasma of patients with SCD. By a series of elegant experiments, they proved that extracellular heme is the principle factor that induces IFN-I. Thus, injection of heme in WT mice resulted in significant, dose-dependent increases in the concentration of IFN-α and -β. Administration of lysate from erythrocytes or hemoglobin also elicited release of IFN-α, but to a lower extent than free heme. Therefore, Liu et al describe a novel immunoregulatory effect of cell-free heme. Because the IFN-I system plays an important role in the defense against viruses, these data raise the question of whether patients with chronic intravascular hemolysis have an increased resistance to many viral infections.
Further, Liu et al demonstrated that the source of IFN-I in SCD (or experimentally after injection of heme) is a population of monocyte-derived macrophages that reside in the liver. The engagement of TLR4 receptor may be a trigger for IFN-I synthesis and release. However, the results demonstrate that heme does not induce expression of IFN-I by engagement of this receptor in TLR4−/− mice. This finding is intriguing, as previous reports demonstrated that heme activates the release of the proinflammatory cytokine TNF-α by mouse macrophages by interaction with TLR4.5 Liu et al found that the use of specific inhibitor of Tank kinase binding 1 and IκB kinase-ε (TBK1/IKKε), amlexanox, curtailed the ability of heme to induce IFN-I by macrophages. How heme activates the signaling pathway resulting in synthesis of IFN-I remains an open question. Is there engagement of some extracellular receptor by heme? Or does heme, by its lipophilic nature, penetrate intracellular membrane and acts within the cell?
In the next part of the investigation, Liu et al focused on the pathophysiological repercussions of heme-induced release of IFN-I. They unambiguously demonstrated that hemolysis-derived heme in SCD mice or externally injected heme causes a significant increase in the recruitment of a specific population of macrophages in the liver. Heme-mediated IFN-I release by liver macrophages triggered differentiation of circulating monocytes to macrophages and their subsequent localization to the liver, increasing their number by sixfold. The main actor in this cascade was the chemokine CCL2 released in response to IFN-I. Notably, Liu et al showed that heme-induced IFN-α significantly augmented the expression of CD64 on the surface of macrophages. A similar effect was observed after in vitro IFN-α exposure of human monocytes from healthy donors or from patients with SCD. CD64 or FcγRIa is a high-affinity receptor for the constant fragment of IgG. Liu et al clearly demonstrated that the increased expression of CD64 resulted in more efficient phagocytosis of erythrocytes opsonized by antibodies. This finding is one of the most important ones in the study. It is particularly relevant for understanding the pathological mechanisms of hemolytic diseases, especially in cases of alloimmunization (eg, development of antibody response against human erythrocytes after repeated transfusions in patients with SCD8). These antibodies can cause a life-threatening reaction referred to as a delayed hemolytic transfusion reaction. Liu et al hypothesized that hemolysis-derived heme and its capacity to induce IFN-I results in an overall decrease in the threshold of antibody concentration necessary for induction of erythrophagocytosis, because of the higher number of macrophages and higher expression of CD64. Thus, lower titers of antibodies would be necessary to trigger erythrophagocytosis. Studies showing that free heme has the capacity to induce polyreactivity of endogenous antibodies9 prompt us to speculate that increased potential for phagocytosis of activated liver macrophages may allow such antibodies to participate in erythrophagocytosis under hemolytic conditions. This possibility offers an explanation about the fact that ∼30% of cases of delayed hemolytic transfusion reaction occur in the absence of typical alloantibodies against red blood cells.
In summary, Liu et al revealed a novel biological effect of extracellular heme. They also uncovered an important pathological mechanism leading to destruction of erythrocytes in hemolytic diseases. Further studies are warranted in this area, which could lead to development of innovative therapeutic strategies for disease associated with release of heme.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal