

Key Points
Multiplex mutagenesis in human hematopoietic stem and progenitor cells is efficient and without evidence of toxicity.
In vivo disruption of BCL11a expression and its binding site within the β-globin locus enhances HbF reactivation in β-thalassemia.

Abstract
Thalassemia or sickle cell patients with hereditary persistence of fetal hemoglobin (HbF) have an ameliorated clinical phenotype and, in some cases, can achieve transfusion independence. Inactivation via genome editing of γ-globin developmental suppressors, such as BCL11A or LRF/ZBTB7A, or of their binding sites, have been shown to significantly increase expression of endogenous HbF. To broaden the therapeutic window beyond a single-editing approach, we have explored combinations of cis- and trans-editing targets to enhance HbF reactivation. Multiplex mutagenesis in adult CD34+ cells was well tolerated and did not lead to any detectable defect in the cells’ proliferation and differentiation, either in vitro or in vivo. The combination of 1 trans and 1 cis mutation resulted in high editing retention in vivo, coupled with almost pancellular HbF expression in NBSGW mice. The greater in vivo performance of this combination was also recapitulated using a novel helper-dependent adenoviral-CRISPR vector (HD-Ad-dualCRISPR) in CD34+ cells from β-thalassemia patients transplanted to NBSGW mice. A pronounced increase in HbF expression was observed in human red blood cells in mice with established predominant β0/β0-thalassemic hemopoiesis after in vivo injection of the HD-Ad-dualCRISPR vector. Collectively, our data suggest that the combination of cis and trans fetal globin reactivation mutations has the potential to significantly increase HbF both totally and on a per cell basis over single editing and could thus provide significant clinical benefit to patients with severe β-globin phenotype.
Introduction
β -globin disorders, or β-hemoglobinopathies, represent the most common monogenic disorders worldwide, with β-thalassemia and sickle cell disease being the most prevalent forms. Despite significant advances on developing novel therapies for hemoglobinopathies, challenges still exist, with a widely applicable and robust therapeutic approach for both sickle cell disease and β-thalassemia still a work in progress. Elevated production of fetal hemoglobin (HbF) throughout adult life, a benign condition known as hereditary persistence of HbF (HPFH), has been shown to ameliorate the clinical and hematologic severity of both conditions.1 Recently, genome-editing approaches have opened up new therapeutic opportunities, enabling specific reactivation of the developmentally silenced HbF. An approach that has gained great momentum over the last few years is the targeted disruption of transcriptional regulatory pathways that suppress HbF expression during development and throughout adulthood. In particular, the regulatory axes of BCL11A and LRF/ZBTB7A, key transcriptional repressors of HbF expression, have emerged as attractive candidates. Although initial attempts focused on perturbing the expression or function of the BCL11A and LRF transcription factors themselves,2-6 the recent discovery of their cognate binding sites within the fetal globin promoters (HBG1/HBG2) has provided additional targets for intervention. Specifically, BCL11A has been shown to bind to the TGACCA motif at −115 in the HBG1/2 promoters,7,8 whereas LRF binds to an element at −200.7 Promisingly, naturally occurring single-nucleotide variants that cluster at these 2 sites are associated with HPFH and indeed, targeted perturbation of these sites by genome engineering by us and others has been shown to result in marked reactivation of HbF expression.9-11 In the present study, we performed simultaneous genome editing of the aforementioned cis (HBG-promoter binding sites) and trans (BCL11A enhancer) target sites to further increase levels of in vivo HbF reactivation and expand applicability of genome editing–mediated therapy to more demanding cases, such as β0/β0-thalassemic patients. Furthermore, we incorporated the most efficient combination into a novel helper-dependent adenoviral vector (HD-Ad5/35++) for in vivo delivery as an inexpensive, efficient, and widely applicable in vivo gene therapy modality for β-hemoglobinopathies.
Methods
Detailed information on methods can be found in supplemental Methods (available on the Blood Web site).
RNP transfection in CD34+ cells
CD34+ cells from mobilized healthy donors were cultured in an optimized culture medium for hematopoietic stem cell (HSC) maintenance and expansion12 for 2 days prior to ribonucleoprotein (RNP) transfection. For the RNP formation, 32 μg of NLS-Cas9 were incubated with the individual single guide RNAs (sgRNAs) in a 1:6 ratio. For all CRISPR combinations, the different RNPs were formed and incubated individually and combined with the cells right before transfection. Transfection was performed in the ECM 830 Square Wave Electroporation System (BTX, Holliston, MA) according to the manufacturer’s instructions. The cells were cultured in the hematopoietic stem and progenitor cell (HSPC) expansion medium for 2 additional days before erythroid differentiation13 and/or transplantation in NBSGW14 mice.
Adenoviral transduction of HUDEP-2 and thalassemic CD34+ cells
HUDEP-2 cells were cultured as previously reported.3 Cells were transduced at various multiplicities of infection (MOIs) with adenoviral vectors in low-attachment plates. Erythroid differentiation was induced in the same basal medium excluding dexamethasone, but in the presence of doxycycline and stem cell factor.
CD34+ cells from patients with β-thalassemia major were collected during 2 mobilization clinical trials conducted at G. Papanikolaou Hospital, Thessaloniki, Greece.15,16 The cells were grown in expansion medium for 1 day prior to transduction with the helper-dependent adenoviral-CRISPR vector (HD-Ad-dualCRISPR; MOI = 4000). The virus was removed after 48 hours and the cells were either cultured under erythroid conditions or plated in methylcellulose base medium for colony evaluation.
Xenotransplantation studies
In all of our xenotransplantation studies, the NBSGW mouse model14 was used. All procedures were approved by the institutional animal care and use committee. For our ex vivo genome-editing experiments, 106 cells were transplanted in nonirradiated mice 48 hours post-RNP transfection. Cell engraftment was evaluated in peripheral blood monthly and multilineage reconstitution was assayed in the mouse bone marrow 16 weeks posttransplantation. At that time, the mice were euthanized; bone marrow was collected and either ex vivo cultured or assayed for hematopoietic lineage constitution, editing levels, and HbF expression.
For our in vivo genome-editing experiments, 2 × 106 CD34+ cells from a β0/β0-thalassemic patient were transplanted in NBSGW mice. After establishment of human hematopoiesis in the transplanted mice (confirmed by fluorescence-activated cell sorting [FACS] at 6 weeks posttransplantation), the mice were mobilized with Plerixafor plus granulocyte colony-stimulating factor (G-CSF) as previously described.17 At the end of mobilization, the mice were injected with the HD-Ad-dualCRISPR vector (4 × 1010 viral particles). The mice were euthanized 16 weeks posttransplantation of CD34+ cells. The size of the collected spleens was evaluated, and their bone marrow was assayed for overall engraftment, multilineage reconstitution, and HbF expression in the human erythroid engrafted cells or in erythroid colonies (burst-forming units erythroid [BFU-Es]).
Statistical analyses
Comparisons between 2 groups were carried out using the Student t test. For comparisons of multiple groups, 1- and 2-way analysis of variance (ANOVA) with Bonferroni posttesting for multiple comparisons was used.
Results
Comparison of HbF-reactivation efficiency in adult normal CD34+ cells by cis and trans genome editing
To test how the therapeutic potential of HBG promoter mutations compares to the broadly deployed BCL11A-enhancer disruption, we explored the effect in primary, human CD34+ HSCs (Figure 1A). The guide RNAs (gRNAs) targeting the BCL11A-enhancer (hereafter BCL11A) and the HBG-115 have already been described elsewhere3,9 (Figure 1B). For the HBG-200 region, we tested 3 gRNAs (HBG-195, -196, -197) in CD34+ cells and selected HBG-197 for follow-up experiments as it achieved the highest editing rate compared with the other 2 (supplemental Figure 1a-b). To assess the specificity of HBG-197 gRNA, we colabeled the 53BP1 protein and an ∼2-kb stretch of the target genomic cut site using a recently developed, novel interphase oligo DNA–fluorescence in situ hybridization (FISH) technique (supplemental Methods; supplemental Figure 1c). We imaged mock and nuclease-treated cells 3-dimensionally and measured the extent of spatial colocalization between 53BP1 and the FISH signal as well as the total 53BP1 foci counts per cell at +3, +6, and +24 hours posttransfection. Three-dimensional (3D) analysis on 750 single cells per condition in HUDEP2 cells showed minimal activity of noncolocalized 53BP1 in nuclease-treated cells relative to mock-treated cells (supplemental Figure 1d-e).

Gene editing of human CD34+ cells after RNP electroporation. (A) Schematic depiction of the genome editing targets in chromosomes 2 and 11. (B) Sequence of the optimal sgRNAs used for the downstream assays. Overlined is the binding sequence of the transcription factors GATA1, LRF, and BCL11A, respectively. (C) Percentage of introduced indels 2 days post-RNP electroporation. (D) Frequency of microhomology-mediated end joining (MMEJ) within the different indels. (E) Most abundant genotypes (representing >2% of the alleles) post–genome editing for the 3 loci. All plots represent data from at least 4 different CD34+ cell donors. Values are represented as means ± standard error of the mean (SEM). ***P ≤ .0001, *P ≤ .05 vs untreated (untr); ††P ≤ .001, †P ≤ .05 vs HBG-197, ‡P ≤ .05 vs HBG-115 (unpaired Student t test).
Gene editing of human CD34+ cells after RNP electroporation. (A) Schematic depiction of the genome editing targets in chromosomes 2 and 11. (B) Sequence of the optimal sgRNAs used for the downstream assays. Overlined is the binding sequence of the transcription factors GATA1, LRF, and BCL11A, respectively. (C) Percentage of introduced indels 2 days post-RNP electroporation. (D) Frequency of microhomology-mediated end joining (MMEJ) within the different indels. (E) Most abundant genotypes (representing >2% of the alleles) post–genome editing for the 3 loci. All plots represent data from at least 4 different CD34+ cell donors. Values are represented as means ± standard error of the mean (SEM). ***P ≤ .0001, *P ≤ .05 vs untreated (untr); ††P ≤ .001, †P ≤ .05 vs HBG-197, ‡P ≤ .05 vs HBG-115 (unpaired Student t test).
G-CSF–mobilized CD34+ cells from healthy donors were then transfected with all 3 RNPs: BCL11A, HBG-197, and HBG-115. Editing levels for all RNPs were above 50%. Specifically, insertions and deletions (indels) were 52.6% ± 3.2% for HBG-197, 72.8% ± 7.02% for HBG-115, and 91.7% ± 2.8% for BCL11A (Figure 1C). Microhomology-mediated end joining was only observed at appreciable levels in BCL11A (11.8% ± 1.3%) and HBG-115 (27% ± 3.9%) (Figure 1D), with the majority of the latter being the naturally occurring −114 to −102, 13-bp HPFH deletion,18 consistent with previous reports9,11,19 (Figure 1E). The overall growth and enucleation of the HBG-edited and BCL11A-enhancer edited cells during erythroid differentiation was identical to that of untreated cells (Figure 2A-B). At the completion of the erythroid differentiation, HbF expression was assayed by both flow cytometry and high-performance liquid chromatography (HPLC). Overall , HbF reactivation between the BCL11A-erythroid enhancer and the HBG-edited samples was similar, and, in all cases, significantly increased over unedited cells. Specifically, BCL11A-enhancer knockout yielded 39.7% ± 10.1% HbF+ red blood cells, whereas HBG-197 and HBG-115 gave 49.2% ± 5.9% and 52.2% ± 6.7% HbF+ red blood cells, respectively, compared with 21.4% ± 2.3% of HbF+ cells in the control culture (Figure 2C). Despite BCL11A editing not yielding the highest number of F cells in the bulk population, it had the highest impact on γ-globin expression (γ/α globin P = .00014 vs untreated, P = .032 and P = .02 for HBG-197 vs untreated and HBG-115 vs untreated, respectively) as assayed by HPLC (Figure 2D), suggesting a higher per cell γ-globin reactivation. Additionally, HPLC revealed a preferential increase in Gγ-globin expression after editing the BCL11A enhancer (Figure 2E-F), whereas both of the HBG promoter disruptions preferentially increased the expression of the Aγ chain (Figure 2E-F). This could be at least partially attributed to dual double-stranded breaks in the duplicated HBG genes, resulting in the excision of HBG2/Gγ, as has been previously described.9,11,19
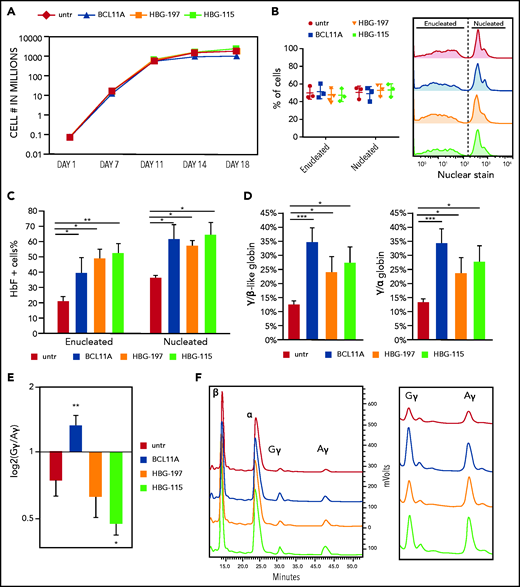
Direct comparison of the 3 RNPs on their effect on erythropoiesis and HbF reactivation. (A) Cell growth over time during in vitro erythroid differentiation. (B) Enucleation of the erythroid cells on day 18 of the differentiation. (C) HbF+ cell frequency as assayed by flow cytometry. (D) HPLC analysis of γ-globin chain expression presented as ratio over the β-like globins and α-globin chain. (E) Ratio of Gγ/Aγ chains postediting with the 3 nucleases. (F) HPLC tracks depicting the difference in Gγ/Aγ balance between the different samples. All plots represent data from at least 4 different CD34+ cell donors. Values are represented as means ± SEM. ***P ≤ .0001, **P ≤ .001, *P ≤ .05 vs untreated (untr) (unpaired Student t test).
Direct comparison of the 3 RNPs on their effect on erythropoiesis and HbF reactivation. (A) Cell growth over time during in vitro erythroid differentiation. (B) Enucleation of the erythroid cells on day 18 of the differentiation. (C) HbF+ cell frequency as assayed by flow cytometry. (D) HPLC analysis of γ-globin chain expression presented as ratio over the β-like globins and α-globin chain. (E) Ratio of Gγ/Aγ chains postediting with the 3 nucleases. (F) HPLC tracks depicting the difference in Gγ/Aγ balance between the different samples. All plots represent data from at least 4 different CD34+ cell donors. Values are represented as means ± SEM. ***P ≤ .0001, **P ≤ .001, *P ≤ .05 vs untreated (untr) (unpaired Student t test).
Multiplex mutagenesis in CD34+ cells
According to the data described above (Figure 2C-D) , BCL11A knockdown appears to have a greater effect in per cell HbF reactivation, whereas HBG-115 or HBG-197 disruption can potentially result in a higher number of F cells. We therefore envisioned that by combining these 2 targets, an additive or synergistic effect may ensue. To this aim, we tested all possible RNP combinations in CD34+ cells (Figure 3A). The editing levels for HBG-115 and BCL11A were similar whether they were multiplexed or not, whereas a slight but not significant increase of indel percentage was observed when multiplexing HBG-197 sgRNA with any other target (Figure 3B). The HBG1 and HBG2 promoters consist of nearly identical sequences, therefore simultaneous cleavage by ≥1 RNPs can result in deletion of the intervening sequence, containing the entire HBG2 gene. We adopted a sequencing method developed by Métais et al19 to evaluate the percentage of HBG2 loss. Similar levels of HBG2 deletion were observed between the HBG-alone and BCL11A-HBG combinations (supplemental Figure 2a). However, HBG2 deletion was significantly lower when multiplexing HBG-115 with HBG-197. By genotyping these samples, we observed that the majority of the occurred genotypes (>70% of the edited alleles) had an 82-bp deletion between the −197 and −115 sites (supplemental Figure 2b). We subsequently evaluated the levels of HbF expression both by FACS and HPLC. Both the HbF+ cell frequency and total γ-lobin levels were significantly increased by combining BCL11A editing with editing of either 1 of the HBG sites (Figure 3C-D), resulting in almost pancellular HbF expression in erythroid cells (Figure 3C). Somewhat expectedly, the 82-bp deletion generated from the combined HBG-115 and HBG-197 editing, resulted in significantly lower HbF levels compared with either mutation alone (Figure 3D), as it depleted a CACCC motif previously described as harboring an HbF activator (SP1/KLF).20 Analysis of the clonogenic capacity of the double-edited cells revealed no significant difference compared with the control groups, as the frequency of generated BFU-E and colony-forming unit granulocyte macrophage (CFU-GM) colonies was similar in all conditions (Figure 3E). To assess DNA damage response (DDR) at a single-cell level after multiplex genome editing, we adopted an immunofluorescence imaging approach21 to assay the per cell load of the DNA repair protein 53BP1. Analysis of 250 individual cells from 3 healthy donors 6 hours posttransfection revealed, for the combination of 2 targets (BCL11A plus HBG-115), an average of 2.9 ± 0.5 53BP1 foci per nucleus, slightly greater than the levels observed for cells edited with individual RNPs (BCL11A, 2.3 ± 0.4; HBG-115, 2.3 ± 0.4; Figure 3F top row) and reflective of simultaneous double-strand breaks at both targets. However, 24 hours posttransfection, the 53BP1 level in all edited cells with either 1 or 2 RNPs was similar to the unedited cells, suggesting absence of persistent mutagenesis after completion of the targeted genome-editing events (Figure 3F, bottom row). Furthermore, to test whether the individual or multiplexed editing approaches generated any aberrant chromosomal translocations between the targeted BCL11a and HBG loci, we adopted a high-resolution 2-color DNA FISH-based imaging approach to check for translocations at the single-cell level. By labeling genomic loci adjacent to the 2 target cut sites in 2 different colors, a translocation event would be detected by a spatial colocalization of the 2 colors in microscope images (supplemental Figure 3a). HUDEP2 cells and CD34+ cells that had fully recovered after gRNA transfection were imaged 3-dimensionally and checked for translocations by measuring the extent of spatial colocalization between the 2 labeled loci. 3D colocalization analysis on 200 single cells per condition in HUDEP2 and CD34+ cells showed minimal colocalization between the BCL11a and HBG loci in all combinations (supplemental Figure 3b-d).
![Multiplex mutagenesis in CD34+ cells to enhance endogenous HbF reactivation. (A) Schematic depiction of the nuclease combinations tested. (B) Editing levels presented as indel percentage in cells edited with 1 (SKO) or 2 (DKO) RNPs. (C) Histogram plots of HbF expression assayed by flow cytometry. (D) γ-globin chain expression presented as fold difference over the untransfected samples (HPLC). (E) Clonogenic capacity of the edited cells with the different RNP combinations. (F) Immunofluorescence imaging of the abundance of 53BP1 to assess DNA damage response and toxicity after introducing 2 mutations in different loci in CD34+ cells 6 and 24 hours postelectroporation. 4′,6-diamidino-2-phenylindole (DAPI; blue)/ 53BP1 (red) stain; scale bar, 5 μm. Values are represented as means ± SEM (n ≥ 3). *P ≤ .05 vs untreated (untr), †P ≤ .05 vs HBG-197, ‡P ≤ .05 vs HBG-115 (2-way analysis of variance [ANOVA]).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/17/10.1182_blood.2020010020/8/m_bloodbld2020010020f3.png?Expires=1769135864&Signature=OEvdLeAJHFEWYiqHsYXOOKD4H4FbJQDyS68kp68TyQYBzv1l2I8~9IUqx5xOeN-10euEyZAz3iWCVFZqXvS5XaAp1TPfgy38v1fAPo6dxgEZODK2KPL9tCWVwsDge1hm4iorcQDNk~03l-Qo4Ka5zYBg0kXJEExIJhCD4vDKIM4E~i9Y8~yOKhpV01-i3nDTLqsY8ABbuGfmNK1diRxE038MGloEo~w7mN6iko47r5UrrWtCS3t695LxBQgkDHGEXU6ZK2LzPpw-iXfD8-9nt5-aKhIlY3DFrUjh9jr3OcHXZ-NfmA6soubB8idZxeBK2gpwpMQJFJE5Aj-pdnLGOg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Multiplex mutagenesis in CD34+ cells to enhance endogenous HbF reactivation. (A) Schematic depiction of the nuclease combinations tested. (B) Editing levels presented as indel percentage in cells edited with 1 (SKO) or 2 (DKO) RNPs. (C) Histogram plots of HbF expression assayed by flow cytometry. (D) γ-globin chain expression presented as fold difference over the untransfected samples (HPLC). (E) Clonogenic capacity of the edited cells with the different RNP combinations. (F) Immunofluorescence imaging of the abundance of 53BP1 to assess DNA damage response and toxicity after introducing 2 mutations in different loci in CD34+ cells 6 and 24 hours postelectroporation. 4′,6-diamidino-2-phenylindole (DAPI; blue)/ 53BP1 (red) stain; scale bar, 5 μm. Values are represented as means ± SEM (n ≥ 3). *P ≤ .05 vs untreated (untr), †P ≤ .05 vs HBG-197, ‡P ≤ .05 vs HBG-115 (2-way analysis of variance [ANOVA]).
Multiplex mutagenesis in CD34+ cells to enhance endogenous HbF reactivation. (A) Schematic depiction of the nuclease combinations tested. (B) Editing levels presented as indel percentage in cells edited with 1 (SKO) or 2 (DKO) RNPs. (C) Histogram plots of HbF expression assayed by flow cytometry. (D) γ-globin chain expression presented as fold difference over the untransfected samples (HPLC). (E) Clonogenic capacity of the edited cells with the different RNP combinations. (F) Immunofluorescence imaging of the abundance of 53BP1 to assess DNA damage response and toxicity after introducing 2 mutations in different loci in CD34+ cells 6 and 24 hours postelectroporation. 4′,6-diamidino-2-phenylindole (DAPI; blue)/ 53BP1 (red) stain; scale bar, 5 μm. Values are represented as means ± SEM (n ≥ 3). *P ≤ .05 vs untreated (untr), †P ≤ .05 vs HBG-197, ‡P ≤ .05 vs HBG-115 (2-way analysis of variance [ANOVA]).
Engraftment of double-edited HSCs in vivo
Because the combinations of BCL11A plus HBG-197 or BCL11A plus HBG-115 edits achieved the highest HbF levels in vitro, we pursued the in vivo evaluation of their engraftability, retention of genome editing and the HbF expression/reactivation in engrafted erythroid cells. To this aim, CD34+ cells were expanded for 2 days in cytokine-enriched medium supplemented with 3 small molecules (StemRegenin1, UM171, Ly2228820) to achieve HSC expansion before transfection as we have previously described.4,12 As controls, we used CD34+ cells that were either untransfected or transfected with any of the 3 single RNPs. Two days posttransfection, the cells were transplanted in a NBSGW mouse model without conditioning. The mice were euthanized 16 weeks posttransplantation (Figure 4A). We did not observe a difference in overall engraftment levels of human CD45+ cells in any of the assayed conditions. Multilineage engraftment was also evident in all mice, with no skewing toward a particular lineage in any of the different groups (Figure 4B). Assessment of the bone marrow HbF percentage expression within the erythroid Glycophorin A (GlyA)+ cells in transplanted mice revealed that single editing of either the BCL11A enhancer or HBG-115 could yield up to 71% F+ cells, whereas editing of the HBG-197 was <50%, but still significantly higher than the control group. The highest number of HbF+ cells was observed in the BCL11A plus HBG-115 combination, reaching up to almost 100% of the engrafted GlyA+ cells in 1 of the mice (Figure 4C). The HbF difference between the single- and dual-edited cells in vivo was more profound when looking at the γ-globin chain expression in the BCL11A plus HBG-115 combination, suggesting higher γ-globin expression per cell. However, as already shown by FACS, HBG-197-RNP alone did not increase HbF expression compared with the control, whereas the signal observed in the BCL11A plus HBG-197 combination was attributed solely to BCL11A editing. The evaluation of editing levels in the bone marrow of engrafted mice revealed retention of the high editing frequency for the BCL11A enhancer with only 15.1% decrease from the input cells, a considerable 43.3% drop in the in vivo–editing levels for the HBG-115 site and a dramatic loss (68,2%) of edited cells in the HBG-197 group, justifying the overall lower number of HbF+ cells in vivo and low γ-globin chain expression (Figure 4E).

In vivo performance of the double-edited cells. (A) Experimental procedure of the in vivo experiments. CD34+ cells from healthy donors were cultured in the presence of cytokines and a small molecule combination to maintain their HSC population for 2 days before electroporation with the respective RNPs or RNP combinations. The cells were transplanted into NBSGW mice 2 days posttransfection. The mice were euthanized 16 weeks posttransplantation and their bone marrow (BM) was collected for further analysis. (B) Engraftment and multilineage reconstitution of the human cells in the recipients 16 weeks posttransplantation. (C) HbF expression in BM-engrafted human erythroid (GlyA+) cells assayed by FACS. (D) HPLC data for γ-globin chain expression. The values are represented as a fold difference from mice receiving untransfected cells. (E) Indel frequency per loci in BM-engrafted human cells (dots) and in the input cells (bars). Each dot represents a different mouse. The indel drop is presented as a percentage on top of the graph. Values are represented as means ± SEM (n ≥ 3 mice). (F) Composition of colonies pre- (in vitro) and post- (in vivo) transplantation in terms of indel presence in 1 or both targeting sites.
In vivo performance of the double-edited cells. (A) Experimental procedure of the in vivo experiments. CD34+ cells from healthy donors were cultured in the presence of cytokines and a small molecule combination to maintain their HSC population for 2 days before electroporation with the respective RNPs or RNP combinations. The cells were transplanted into NBSGW mice 2 days posttransfection. The mice were euthanized 16 weeks posttransplantation and their bone marrow (BM) was collected for further analysis. (B) Engraftment and multilineage reconstitution of the human cells in the recipients 16 weeks posttransplantation. (C) HbF expression in BM-engrafted human erythroid (GlyA+) cells assayed by FACS. (D) HPLC data for γ-globin chain expression. The values are represented as a fold difference from mice receiving untransfected cells. (E) Indel frequency per loci in BM-engrafted human cells (dots) and in the input cells (bars). Each dot represents a different mouse. The indel drop is presented as a percentage on top of the graph. Values are represented as means ± SEM (n ≥ 3 mice). (F) Composition of colonies pre- (in vitro) and post- (in vivo) transplantation in terms of indel presence in 1 or both targeting sites.
To confirm that cells with multiplexed mutations were able to engraft, we evaluated colony-forming cells from the mouse bone marrow edited in either the HBG promoter or BCL11A enhancer site. We failed to detect any colony with mutations in both BCL11A plus HBG-197 sites, consistent with the dramatic in vivo loss of editing observed at the HBG-197 site (Figure 4F). However, for the BCL11A plus HBG-115 combination, >70% of the bone marrow–derived colonies were marked with mutations on both sites, a frequency almost identical to that observed in the preengraftment population (Figure 4E-F).
Developing a therapeutic vector for multiplex mutagenesis
We have recently developed a novel gene therapy approach that overcomes the difficulties of cell collection, ex vivo cell modification and conditioning.22-24 In this approach, a new-generation HDAd5/35++ CRISPR vector is used to efficiently edit HSPCs either in vitro or in vivo.10,25 We have previously demonstrated that in vivo HDAd5/35++ CRISPR injection results in disruption of the HBG promoter within the BCL11A-binding site, leading to a pronounced switch from human β- to γ-globin expression in red blood cells of adult β-YAC mice that was maintained even in secondary recipients.10 We therefore decided to modify this vector and introduce a second sgRNA targeting the BCL11A enhancer (Figure 5A). The newly generated adenoviral vector for dual genome editing (Ad-dualCRISPR) was first tested in human-derived erythroid progenitor cells (HUDEP-2), and vectors including only 1 of the 2 sgRNAs were used as controls (Figure 5A). No effect on cell viability, proliferation, and maturation was observed after transduction with various MOIs of the Ad-dualCRISPR (supplemental Figure 4). Deep sequencing of the Ad-dualCRISPR vector-infected cells showed a target site-specific indel frequency of 76.6% for the BCL11A-enhancer target site and 72.7% for the HBG promoter target site (supplemental Figures 5 and 6). At the end of the erythroid differentiation/maturation of HUDEP-2 cells, we observed >60% HbF+ cells in the Ad-dualCRISPR vector-transduced, ∼18% in Ad-sgBCL11A vector-transduced, and ∼50% in Ad-sgHBG vector-transduced cells as compared with ∼2% HbF+ cells in untransduced cells (Figure 5B-C). A significant difference in γ-globin expression was also demonstrated by HPLC of cell lysates among different vectors (Figure 5D). To investigate the occurrence of both mutations in single cells, we clonally expanded cells edited with either of the 3 vectors and analyzed the clones for editing and HbF expression by flow cytometry. Importantly, the average HbF expression level (mean fluorescent intensity [MFI]) in clones derived from the Ad-dualCRISPR–transduced cells was significantly higher than in clones from Ad-BCL11A– or Ad-HBG–infected cells (Figure 5E), indicating that introduction of both mutations increase γ-globin expression per cell in addition to the increase in the population level. Analysis of the HBG2 gene deletion in 50 clones showed that 42% and 32% had biallelic and monoallelic deletions of the 4.9-kb region, respectively (supplemental Figure 7a-b). In HBG-115–edited clones, the HbF MFI was similar irrespective of the presence of intact HBG2 or its biallelic or monoallelic deletion. However, in double-edited clones, HbF MFI was synergistically increased only when both copies of HBG2 were present, indicating a possible requirement of an intact HBG2 gene for the synergistic effect of the 2 mutations (supplemental Figure 7c).
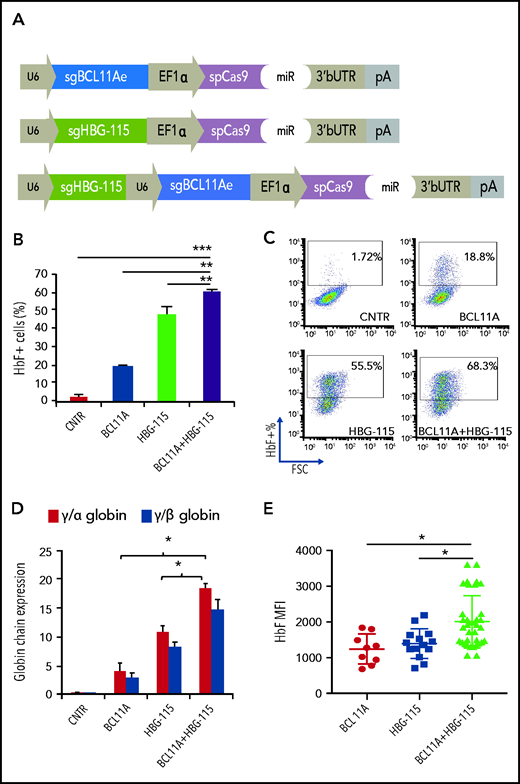
An HD-Ad5/35++ vector for multiplex mutagenesis in human erythroid cells. (A) Design of the 3 adenoviral vectors used in downstream experiments. (B) HbF expression in bulk-transduced HUDEP-2 cells after transduction with the different Ad-CRISPRs. All samples were transduced with the same MOI. (C) Representative HbF FACS dot plots. (D) γ-globin expression as evaluated by HPLC. (E) HbF expression presented as mean fluorescent intensity (MFI) in single-cell clones derived from Ad-BCL11A–, Ad-HBG-115–, or Ad-BCL11A + HBG-115–transduced cells. Values are represented as means ± SEM. ***P ≤ .0001, **P ≤ .001, *P ≤ .05 (unpaired Student t test).
An HD-Ad5/35++ vector for multiplex mutagenesis in human erythroid cells. (A) Design of the 3 adenoviral vectors used in downstream experiments. (B) HbF expression in bulk-transduced HUDEP-2 cells after transduction with the different Ad-CRISPRs. All samples were transduced with the same MOI. (C) Representative HbF FACS dot plots. (D) γ-globin expression as evaluated by HPLC. (E) HbF expression presented as mean fluorescent intensity (MFI) in single-cell clones derived from Ad-BCL11A–, Ad-HBG-115–, or Ad-BCL11A + HBG-115–transduced cells. Values are represented as means ± SEM. ***P ≤ .0001, **P ≤ .001, *P ≤ .05 (unpaired Student t test).
Induction of HbF and amelioration of phenotype in thalassemic erythroid–edited cells
We subsequently tested the efficiency of the Ad-dualCRISPR in CD34+ HSCs from thalassemic patients (n = 3; supplemental Table 1), previously enrolled in mobilization trials.16,26 Following a 48-hour transduction, the cells were cultured in methylcellulose and erythroid differentiation medium and the efficacy of genome editing and extent of γ-globin induction were evaluated. Notably, the double editing did not alter the frequency of hematopoietic stem cells (CD34+/CD38-/CD90+ cells) or their multilineage colony-forming potential (Figure 6A-B). During erythroid differentiation, transduced thalassemic cells proliferated similarly to the untransduced cells (supplemental Figure 8a), however, the percentage of erythroid cells (GlyA+) was significantly higher after Ad-dualCRISPR transduction at all time points to the end of differentiation (supplemental Figure 8b). Moreover, at the end of the culture, there was a highly homogenous GlyA+ population (>95%) that had originated from the Ad-dualCRISPR–transduced CD34+ cells (Figure 6C). This was in stark contrast to the significantly lower and variable frequency of GlyA+ cells (ranging between 30% and 98%), derived from the untransduced, thalassemic CD34+ cells, suggestive of defective erythroid maturation (Figure 6C). Consistent with this, we observed an erythroid maturation blockade in unedited/untransduced cells, which differed markedly from the normal erythroid differentiation and higher enucleation rates of the edited/transduced thalassemic cells (Figure 6D-E).
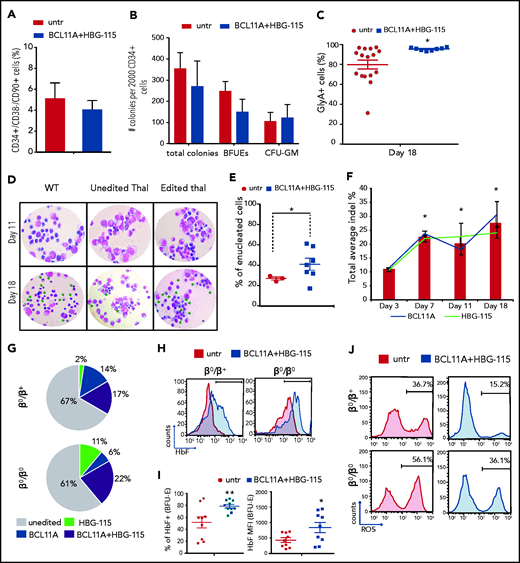
Multiplex mutagenesis in CD34+ cells from patients with β-thalassemia major. (A) Frequency of CD34+/CD38−/CD90+ cells 2 days post–Ad-dualCRISPR transduction. (B) Number of total, erythroid (BFU-E), and myeloid (CFU-GM) colonies per 2000 CD34+ cells. (C) Percentage of erythroid cells (GlyA+) at the end point of in vitro differentiation. (D) Morphology of unedited normal (wt), unedited thalassemic, and edited thalassemic cells on days 11 and 18 of the differentiation. Green arrows point to late-stage maturing erythroid cells . Hematoxylin & eosin stain; original magnification ×40. (E) Percentage of enucleated RBCs at the end of differentiation. (F) Indel percentage at different time points during differentiation. (G) Composition of the thalassemic progenitor cells (CFCs) in terms of single BCL11A (solid blue), single HBG-115 (solid green), or double BCL11A+HBG-115 (striped) mutations. Unedited colonies are represented with gray. (H) HbF expression in enucleated and nucleated cells. (I) ROS levels of edited and unedited thalassemic cells on day 14 of the differentiation. (J) Representative FACS plots of ROS expression in thalassemic erythroid cells before and after dual editing. Values are represented as means ± SEM. **P ≤ .001, *P ≤ .05 vs untreated (C,E,I) or day 3 (F) (unpaired Student t test).
Multiplex mutagenesis in CD34+ cells from patients with β-thalassemia major. (A) Frequency of CD34+/CD38−/CD90+ cells 2 days post–Ad-dualCRISPR transduction. (B) Number of total, erythroid (BFU-E), and myeloid (CFU-GM) colonies per 2000 CD34+ cells. (C) Percentage of erythroid cells (GlyA+) at the end point of in vitro differentiation. (D) Morphology of unedited normal (wt), unedited thalassemic, and edited thalassemic cells on days 11 and 18 of the differentiation. Green arrows point to late-stage maturing erythroid cells . Hematoxylin & eosin stain; original magnification ×40. (E) Percentage of enucleated RBCs at the end of differentiation. (F) Indel percentage at different time points during differentiation. (G) Composition of the thalassemic progenitor cells (CFCs) in terms of single BCL11A (solid blue), single HBG-115 (solid green), or double BCL11A+HBG-115 (striped) mutations. Unedited colonies are represented with gray. (H) HbF expression in enucleated and nucleated cells. (I) ROS levels of edited and unedited thalassemic cells on day 14 of the differentiation. (J) Representative FACS plots of ROS expression in thalassemic erythroid cells before and after dual editing. Values are represented as means ± SEM. **P ≤ .001, *P ≤ .05 vs untreated (C,E,I) or day 3 (F) (unpaired Student t test).
The editing levels for the HBG-115 and BCL11A sites were similar at the end of the erythroid differentiation at ∼30%, as revealed by deep sequencing. However, on day 7 of erythroid differentiation, we observed a significant increase of the percentage of both BCL11A and HBG-115–editing events compared with day 3, suggesting a selective advantage of the corrected cells (Figure 6F). Evaluating the editing frequency in single-transduced BFU-Es from the thalassemic cells, we observed overall 30% to 40% edited colonies, with the majority of them edited in both target sites (Figure 6G). These editing levels were associated with a significant increase in the frequency of HbF+ cells in enucleated erythrocytes (Figure 6H) as well as in the HbF% of expression and MFI in erythroid colonies (Figure 6I), leading to a significant reduction of ROS levels (Figure 6J). Finally, no chromosomal rearrangements were detected by G-banding karyotyping in double-edited cells during erythroid differentiation, indicating that large genomic translocations did not occur between the 2 edited sites (supplemental Figure 8c).
In vivo transduction/editing of xenografted, human CD34+ thalassemic cells with the AD-dualCRISPR vector
We have previously developed a minimally invasive and clinically translatable approach for in vivo HSPC gene therapy. This method involves mobilization with G-CSF/AMD3100 and IV delivery of a nonintegrating, helper-dependent adenoviral HDAd5/35++ CRISPR vector, displaying high affinity to the hCD46 receptor, which is expressed uniformly on HSPCs. Here, we developed a xenograft model of human thalassemic hemopoiesis to test the in vivo transduction with the Ad-dualCRISPR vector. To this end, NBSGW mice were firstly transplanted with CD34+ cells from a patient with a β0/β0 genotype to establish human thalassemic hemopoiesis. Six weeks posttransplantation, the mice were successfully mobilized with an established mobilization scheme15,17,26 (supplemental Figure 9) and then injected with the HSPC-tropic Ad-dualCRISPR, which selectively transduces the human cells (Figure 7A). At euthanization, 9 weeks post in vivo xenotransduction/editing (16 weeks posttransplantation), human hemopoiesis predominated in the chimeric bone marrow (hCD45+ cells, >65%) (Figure 7B). Multilineage (CD34+, CD19+, CD33+, CD3+) engraftment was evident and similar between the Ad-dualCRISPR-treated and untreated mice (Figure 7B). Unfortunately, genotyping of the engrafted human hematopoietic cells was technically unsuccessful due to DNA degradation. Importantly though, the in vivo gene-edited β0 thalassemic erythroid cells contained ∼50% HbF+ cells as compared with 17.9% in their untreated counterparts (Figure 7C) and the total human γ/β-globin mRNA expression increased by >10-fold (Figure 7D). This efficient HbF reactivation resulted in an improved erythroid lineage maturation profile in vivo (Figure 7E), reduced ROS levels (Figure 7F), and spleen size (Figure 7G). Subsequently, we performed secondary transplantation experiments with cells collected from the bone marrow of the injected primary recipients. Engraftment of human CD45+ thalassemic cells was lower compared with the primary recipients, as expected in all secondary transplantation assays, but similar between the 2 groups in their bone marrow and spleen (Figure 7F). Interestingly, the 2 erythroid compartments this time were comparable in both tissues, in contrast to the primary recipients (Figure 7F). Despite the fact that the corrected cells in this nonthalassemic environment did not show a selective advantage, the difference in HbF expression in hCD235a+ cells in vivo and after ex vivo differentiation of bone marrow cells was maintained (Figure 7I).
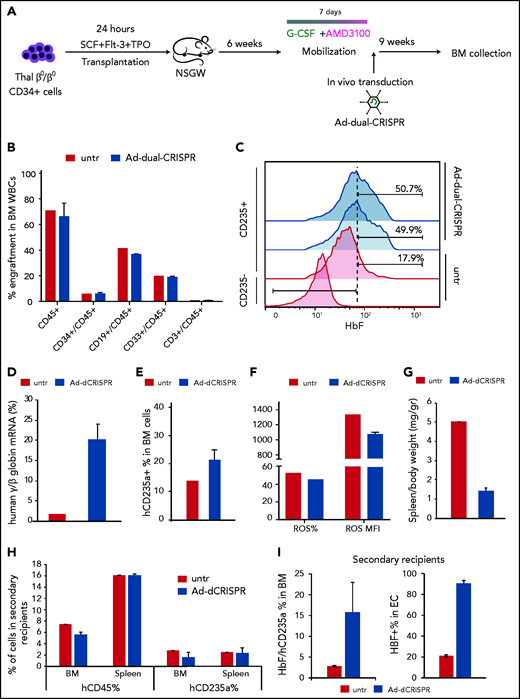
In vivo genome editing of β0/β0-thalassemic cells. (A) Experimental procedure for the in vivo genome-editing experiments. Briefly, mobilized CD34+ cells from a β0/β0-thalassemic patient were transplanted into nonmyeloablated NBSGW mice (n = 3). Six weeks posttransplantation, the mice were mobilized for 7 days with G-CSF + AMD3100. On the seventh day of mobilization, the mice were injected with the Ad-dualCRISPR (Untr = saline). The mice were euthanized 9 weeks later and their BM was collected for further analysis. (B) Multilineage reconstitution 16 weeks posttransplantation. (C) HbF expression in human engrafted nonerythroid (used as control) and erythroid cells assayed by FACS. (D) γ-/β-globin percentage assayed by quantitative RT-PCR. (E) Engraftment of human erythroid cells (GlyA+/CD45−) in the mouse BM at the time of euthanization. (F) Reactive oxygen species (ROS) of the thalassemic engrafted cells at the time of euthanization. (G) Adjusted to weight spleen sizes at the time of euthanization. (H) Engraftment of thalassemic hCD45+ and hCD2345a+ cells in secondary recipients in the BM and spleen as a percentage over the total number or cells. (I) HbF expression in engrafted erythroid cells in the secondary recipients’ BM (left panel) and after ex vivo differentiation of BM cells (right panel).
In vivo genome editing of β0/β0-thalassemic cells. (A) Experimental procedure for the in vivo genome-editing experiments. Briefly, mobilized CD34+ cells from a β0/β0-thalassemic patient were transplanted into nonmyeloablated NBSGW mice (n = 3). Six weeks posttransplantation, the mice were mobilized for 7 days with G-CSF + AMD3100. On the seventh day of mobilization, the mice were injected with the Ad-dualCRISPR (Untr = saline). The mice were euthanized 9 weeks later and their BM was collected for further analysis. (B) Multilineage reconstitution 16 weeks posttransplantation. (C) HbF expression in human engrafted nonerythroid (used as control) and erythroid cells assayed by FACS. (D) γ-/β-globin percentage assayed by quantitative RT-PCR. (E) Engraftment of human erythroid cells (GlyA+/CD45−) in the mouse BM at the time of euthanization. (F) Reactive oxygen species (ROS) of the thalassemic engrafted cells at the time of euthanization. (G) Adjusted to weight spleen sizes at the time of euthanization. (H) Engraftment of thalassemic hCD45+ and hCD2345a+ cells in secondary recipients in the BM and spleen as a percentage over the total number or cells. (I) HbF expression in engrafted erythroid cells in the secondary recipients’ BM (left panel) and after ex vivo differentiation of BM cells (right panel).
Discussion
Genome editing has emerged as a new therapeutic modality to modify candidate suppressor or activator regions within the globin locus for the activation of the developmentally silenced γ-globin. In contrast to lentiviral-mediated gene-addition approaches, genome editing represents a “hit-and-run” approach not requiring lifelong expression of a transgene. Recently, BCL11A and LRF were identified as 2 major repressors of fetal globin in adult life, and knocking down both genes in an erythroid cell line can lead to >90% γ globin as a proportion of total β-like globin expression.6 Repression of either LRF or BCL11A expression is not a clinically viable option because both transcription factors are required for normal physiological function in hematopoietic cells beyond the erythroid lineage.27-31 However, erythroid-specific disruption of BCL11A has been an attractive therapeutic target. Furthermore, BCL11A and LRF act by binding in discrete regions within the γ-globin promoters in sites enriched for natural occurring HPFH mutations,7,8 offering alternative targets to the coding regions of the 2 suppressors. Disruption of any of these 3 loci, the BCL11A enhancer and the 2 sites within the HBG promoters, by either ZFNs,2,4 TALENs,11 or CRISPR-Cas9,3,5,9,19,32 has been linked to an increased number of F cells. To further expand the therapeutic efficacy, especially to more demanding cases where a combination of high numbers of F cells and high HbF expression per cell is required,33 combinatorial approaches have been previously considered and supported by in vitro studies.34,35
Following along that theme, in the present study, we explored a potential additive effect by simultaneous editing of the LRF and BCL11A HBG-binding sites or the combined editing of each binding site with the BCL11A enhancer both in vitro and in vivo. Multiplex mutagenesis has recently moved from plant and mouse studies to primary T cells,36-39 however, no reports of multiplex genome editing in HSCs are available to our knowledge. Here, we showed that the introduction of multiple mutations in different loci and chromosomes in CD34+ cells is highly efficient and not deleterious to the cells as shown by lack of translocation events, but more importantly by the in vivo maintenance of the edited progenitor and stem cell populations with double editing. Even though our imaging-based observations of minimal off-target activity and translocation events postediting may not be as quantitative or as sensitive as deep sequencing–based assays to identify rare (<1%) deleterious events, they do provide an important benefit of measuring the cell-cell variability in single/dual editing by their inherent single-cell resolution. Thus, convincing trends observed from our assays strongly advocate the promise of our editing strategy for clinical translation.
Simultaneous editing of the 2 HBG sites led to significantly lower HbF reactivation compared with either site alone. Analysis of the resulting sequences in the HBG-197 + 115 edited cells revealed that in their majority, the region spanning the exact −115 to −197 positions was missing, thus deleting a known SP1/KLF HBG activator-binding site at −141.20,40 In contrast to the 2 cis mutations, combined editing of the BCL11A enhancer with either HBG site resulted in significantly higher levels of globin expression in vitro compared with either of the nucleases alone, either due to a higher number of total editing events, or due to an additive effect of the 2 nucleases in a proportion of cells. Additional experiments with clonal analysis of the respective genotype in correlation with HbF expression should be performed to uncover the contribution of these alternative mechanisms.
Comparing the different sgRNAs in xenotransplantation experiments, we observed a high retention of genome editing in the BCL11A enhancer by the CRISPR/Cas9 system at similar or higher levels than what we had previously reported by ZFN editing, and in line with recent studies in rhesus monkeys.41 A mild loss of edited cells at the HBG-115 locus was also observed with the majority of the lost alleles containing the microhomology-mediated 13-bp deletion. Surprisingly, we noted a dramatic loss of editing events at HBG-197 (>60%). In a recent study, Weber et al also report a ∼50% loss of indels in HBG-197–edited engrafted cells, which was not marked by the loss of any specific genotype.32 The high in vivo retention of cells edited in the BCL11A enhancer and its HBG1/2-binding site was observed in the double-edited cells as well, as the percentage of human colonies bearing both mutations in the mouse bone marrow was almost identical to that of the input cells. This result shows that multiplex genome editing in HSCs is not detrimental to their in vivo survival. To expand on these results, we used a newly developed, efficient genome-editing technique to simultaneously deliver the 2 gRNAs. Over the last few years, we have been developing a new platform to simplify gene therapy for hemoglobinopathies and make it globally accessible, by in vivo–transducing mobilized HSCs using an HSPC-tropic HD-Ad-5/35++ vector for either gene addition22,23,42 or gene editing.10,25,43 HD-Ad5/35++/CRISPR vectors having large insert capacities, and being nonintegrating, allow safe multiplexing. We therefore modified the previously used HD-Ad-CRISPR targeting the HBG1/2 promoter by adding another sgRNA, geared toward the disruption of the erythroid enhancer of BCL11A. Editing of the 2 sites in mobilized peripheral blood CD34+ cells from thalassemic patients resulted in a significant increase in HbF+ cells, efficient erythropoiesis, higher enucleation rate, and significant reduction of ROS levels, compared with the untreated cells. These effects were markedly improved compared with what we had previously observed by ZFN-mediated BCL11A-enhancer disruption alone.4
Here, we provide for the first time an example of successful in vivo gene therapy in a thalassemic xenotransplant mouse model, showing that double editing of a repressor-binding site and the BCL11A enhancer yields >35% of thalassemic human HBF-expressing cells in the bone marrow and an increased level of HbF per cell. Importantly , these expression levels in both primary and secondary recipients were reached without in vivo selection as in our previous reports (albeit in different models) in which the in vivo selection was critical to obtain a clinically meaningful HBF increase.23,24,43,44 In the present study, selection of the edited cells in vivo was not required, probably due to the survival advantage of the HbF-derepressed HSPCs within the chimeric, predominantly thalassemic, bone marrow. Beyond efficiency, it is acknowledged that our in vivo delivery approach might face immunogenicity challenges, which can be mitigated by the future development of only the Ad35 vector, characterized by very low seroprevalence compared with Ad5.45,46 Collectively, the current study presents, for first time, multiplex gene editing as a method to significantly enhance HbF reactivation, providing clinically relevant levels of engraftment and HbF expression after in vivo HSC gene editing. In vivo multiplex genome editing will potentially overcome several critical obstacles to a wide application of gene therapy for hemoglobinopathies, such as the requirement for leukapheresis, ex vivo manipulation, repeat freezing/thawing of cells, myeloablation, and HSPC transplantation.
Acknowledgments
The authors thank Tobias Ragoczy and William Kerwin for their contribution to the imaging assays.
This work was supported by National Institutes of Health grant R01DK101328-01A1 from the National Institute of Diabetes and Digestive and Kidney Diseases (T.P.) and grant R01HL128288-01 from the National Heart, Lung, and Blood Institute (A.L.). V.N. was supported in part by National Institutes of Health grant RM1-HG007743-02 from the National Human Genome Research Institute Centers of Excellence in Genomic Science Center for Photogenomics. A.G. was financed by both Greece and the European Union (European Social Fund [ESF]) through the Human Resources Development, Education and Lifelong Learning Operational Program in the context of the Strengthening Human Resources Research Potential via Doctorate Research Project (MIS-5000432), implemented by the State Scholarships Foundation (ΙΚΥ). N.P. was supported by the Cooley’s Anemia Foundation and the American Society of Hematology Scholar Award.
Authorship
Contribution: N.P. and A.L. conceptualized the study; N.P., A.L., E.Y., and T.P. designed the experiments; N.P., A.G., C.L., V.N., G.G., R.A., K.P., and A.A. performed the research and interpreted and analyzed the data; G.G., J.N., and D.C. performed genomic and bioinformatic analyses; N.P. and G.G. visualized the study; N.P. wrote the original draft of the manuscript; N.P., A.P.W.F., A.L., E.Y., and T.P. reviewed and edited the manuscript; N.P., A.G., A.L., and T.P. acquired funding; and A.K., A.P.W.F., A.L., E.Y. and T.P. supervised the study.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Thalia Papayannopoulou, Department of Medicine/Hematology, University of Washington, 1705 NE Pacific, Box 357710, Seattle, WA 98195-7710; e-mail: thalp@uw.edu.
Data can be shared through correspondence to either the first or senior authors of the paper at npsatha@altius.org, eyannaki@uw.edu or thalp@uw.edu.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal