

In this issue of Blood, 1 report the successful targeting JAK2 for degradation in cytokine receptor–like factor 2 (CRLF2)-rearranged acute lymphoblastic leukemia (ALL).
CRLF2-rearranged ALL represents 50% of poor prognosis Philadelphia (Ph)-like ALLs. In Ph+ ALL, which harbors BCR-ABL, JAK inhibitors were first shown to restore imatinib sensitivity in mouse models of BCR-ABL–induced ALL, suggesting JAKs may play a role in Ph+ ALLs.2 More recently, Ph-like ALLs were shown to harbor rearranged CRLF2 and also activating mutations in JAK2, such as JAK2 R683G and others, thus pointing to the JAK-STAT pathway as the culprit.3,4
CRLF2 is a subunit bound to JAK2 of the thymic stromal lymphopoietin (TSLP) receptor complex that also contains the IL7 receptor α (IL7R) bound to JAK1.5 Because CRLF2 utilizes JAK2 and JAK2 mutants are detected in these ALLs, an effective therapeutic strategy should be to inhibit JAK2. Therefore, the strategy chosen was to target JAK2 for degradation by modifying ruxolitinib and baricitinib with a linker and molecules known to mediate proteolysis and to form proteolysis-targeting chimeras (PROTAC). These are bifunctional molecules consisting of a ligand that binds an E3 ligase subunit,6 such as cereblon (CRBN), connected by a linker to another small molecule that binds to the target protein, leading to polyubiquitination and proteasomal degradation.6
To design PROTACs, the authors first determined the 3-dimensional crystal structures of ruxolitinib and baricitinib in complexes with the kinase domain of JAK2. These structures are groundbreaking given that these inhibitors are in wide use for hematological, autoimmune, and even severe COVID-19 treatment (baricitinib). The X-ray crystal structures were deciphered previously for ruxolitinib bound to c-Src7 and baricitinib bound to BMP2-inducible kinase (a member of the Numb-associated kinase family),8 whereas the structure of the kinase domain of JAK2 was previously resolved in a complex with a pan-JAK inhibitor,9 but not with the main inhibitory molecules. The new structures established that the C2 carbon of the pyrimidine ring present in both inhibitors is solvent exposed, whereas 2 N atoms of the pyrimidine ring mediate hydrogen bonding interactions with the main-chain amino and carbonyl groups of L932 and E930 residues, respectively, located in the hinge between the N- and C-terminal lobes of JAK2 kinase domain. A linker was attached to the accessible C2 carbon of the pyrimidine ring (see figure). To the linker, either pomalidomide or thalidomide was added to allow CRBN binding. Further chemical modifications were made to reduce the size and enhance cell permeability. A series of >60 compounds was generated, and detailed results are shown for 4 of them, specifically, compounds 5, 6, 7, and 8. Using a kinome assay, the authors show that PROTAC modification leads to loss of tight specificity for ruxolitinib and baricitinib, which are normally JAK1/JAK2 specific. In addition to the 4 JAKs, PROTACs gained specificity for YSK4, MAP3K2, and MAP3K3 and other kinases.
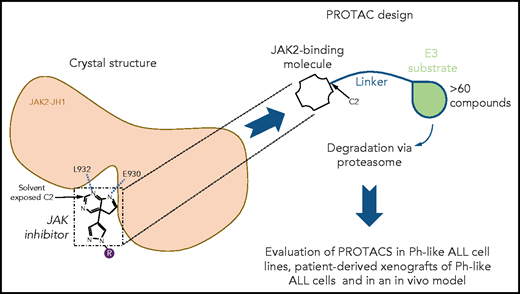
Structure-based design of novel JAK2 degraders. Guided by the novel crystal structures of JAK2 kinase domain bound to type I JAK1/JAK2 inhibitors ruxolitinib and baricitinib, PROTACs were designed by modifying these JAK2 type I ATP competitor inhibitors with a linker and pomalidomide or thalidomide known to mediate PROTAC-based degradation. The structures established that the C2 carbon of the pyrimidine ring present in both inhibitors is solvent exposed allowing attachment of the linker without impairing kinase binding. Furthermore, 2 N atoms of the pyrimidine ring mediate hydrogen bonding interactions with the L932 and E930 residues, in the hinge between the N- and the C-terminal lobes of JAK2 kinase domain. Specificity, degradation of target molecules, and efficacy were assessed in vitro in cell lines and in patient-derived xenografts of Ph-like ALL cells and in an in vivo model using Ph-like ALL cells from a CRLF2 rearranged and JAK2 nonmutated Ph+-ALL patient.
Structure-based design of novel JAK2 degraders. Guided by the novel crystal structures of JAK2 kinase domain bound to type I JAK1/JAK2 inhibitors ruxolitinib and baricitinib, PROTACs were designed by modifying these JAK2 type I ATP competitor inhibitors with a linker and pomalidomide or thalidomide known to mediate PROTAC-based degradation. The structures established that the C2 carbon of the pyrimidine ring present in both inhibitors is solvent exposed allowing attachment of the linker without impairing kinase binding. Furthermore, 2 N atoms of the pyrimidine ring mediate hydrogen bonding interactions with the L932 and E930 residues, in the hinge between the N- and the C-terminal lobes of JAK2 kinase domain. Specificity, degradation of target molecules, and efficacy were assessed in vitro in cell lines and in patient-derived xenografts of Ph-like ALL cells and in an in vivo model using Ph-like ALL cells from a CRLF2 rearranged and JAK2 nonmutated Ph+-ALL patient.
The different PROTACs were then used in cytotoxicity and functional assays in ALL cell lines and xenograft cell lines from several Ph-like ALL patients. Unlike ruxolitinib, which failed to inhibit growth, compounds 5, 6, 7, and 8 induced complete inhibition at concentrations <100 nM. Toxicity in ALL cell lines correlated with the presence of rearranged IGH-CRLF2 and JAK2 mutant cells. Compound 8 was the best. Protein degradation was measured by western blots and the specificity varied among compounds between JAKs and the 2 targets of CRBN, namely G1 to S phase transition 1 (GSPT1), which is involved in effective translation termination of nascent protein chains and the transcription factor IKZF1 (IKAROS family zinc finger 1). The significance of the degradation of this transcription factor in the tumor remains to be determined in vivo. In addition, whether baricitinib-derived PROTACs retain the ability to regulate Numb-associated kinases will also be of interest.
The activity of PROTACs appears to rely on a combination of the on-target effect on JAK2 and, for some of the compounds, the off-target effects of GSPT1. In the MHH-CALL-4 cell line modified to harbor a GSPT1 mutant (G575N) that is not sensitive to CRBN-mediated degradation, the effect of compound 6 was reduced, but the effect of compound 8, which does not target GSPT1, was not affected. Also, in MHH-CALL-4 cells, compounds 5, 6, and 7 inhibited JAK2-STAT5 phosphorylation, and this was more evident when cells were stimulated with TLSP. Compound 7 was assessed in an in vivo system where human primary CRLF2 rearranged/JAK2 wild-type ALL cells expressing YFP, and firefly luciferase were injected in immunodeficient mice. Compound 7 led to degradation of JAK1, JAK2, JAK3, TYK2, and GSPT1, as shown by western blots, and accordingly, led to a significant, but not spectacular, reduction of tumor growth. The tumor reduction in vivo occurred mainly in the peripheral blood and spleen and only to a lower extent in the marrow. Circulating tumor cells may require higher JAK2 signaling levels, or bone marrow penetration may play a role in this tropism.
Last, but not least, ex vivo assessment was performed on a series of xenograft cell lines with different mutations using the 4 compounds (5, 6, 7, 8) vs JAK2 inhibitors (CHZ868, ruxolitinib, and baricitinib) or the thalidomide analog lenalidomide. Interestingly, compound 8 is JAK2 specific and showed good results in some, but not all, xenograft cell lines from ALL patients. Stronger inhibition was exerted by the other compounds, which also degrade GSPT1.
The results presented here provide a pathway forward for the use of PROTACs against JAK-mutated malignancies and offer a new perspective on how current inhibitors can be rationally modified. This approach may be relevant for several subtypes of ALL, where multiple activating mutations in the IL7R and CRLF2 have been reported that lead to activation of JAK1/JAK2.10 Combining this approach with targeting other key events in oncogenesis holds real potential for engineering new therapies against some of the most difficult to treat malignancies.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal