

TO THE EDITOR:
GATA2 deficiency is an autosomal dominant disorder predisposing to myeloid neoplasia and immunodeficiency.1-4 GATA2-related myelodysplastic syndrome (MDS) may present early in life and is commonly associated with monosomy 7 and trisomy 8 karyotypes.5,6 Der(1;7)(q10;p10), henceforth der(1;7), is an unbalanced whole-arm chromosomal translocation resulting in trisomy 1q and deletion 7q (del(7q)) (Figure 1A).7 This translocation has been recurrently reported in adults with primary and therapy-related hematopoietic malignancies, whereas in children it is associated with primary MDS.6,8-10 Previously, we and others reported single cases of GATA2 deficiency that carry der(1;7).4,6,11–15 Building on this intriguing observation, we aimed to define the prevalence of der(1;7) in pediatric MDS according to germline GATA2 mutation (GATA2mut) status and describe the features of this unique subgroup. We evaluated 1620 children and adolescents with primary MDS consecutively enrolled in the registries of the European Working Group of MDS in Childhood (NCT00047268, NCT00662090) with reference review of cytogenetic data. Der(1;7) was present in 13 patients, accounting for 0.8% of the cohort (Figure 1B). In comparison, prior studies mainly in adult MDS reported der(1;7) with a frequency ranging from 1% to 3%.10,16,17 We performed sequencing of GATA2 coding regions and intron 4 (NM_032638.4) and found pathogenic germline GATA2mut in 8 of 11 evaluated der(1;7) cases (Figure 1B). To examine the distribution of der(1;7) according to GATA2mut status, we sequenced 811/1620 pediatric patients with MDS with available specimens. Among the 811 cases, 87 had germline GATA2mut. We observed significant enrichment of der(1;7) in GATA2mut compared with the GATA2 wild type (GATA2wt) patients (9.2%, 8/87 vs 0.4%, 3/724, P < .0001). Of note, we did not observe isolated del(7q) in the GATA2mut cohort, and the only other chromosome 7 aberration was complete monosomy 7.
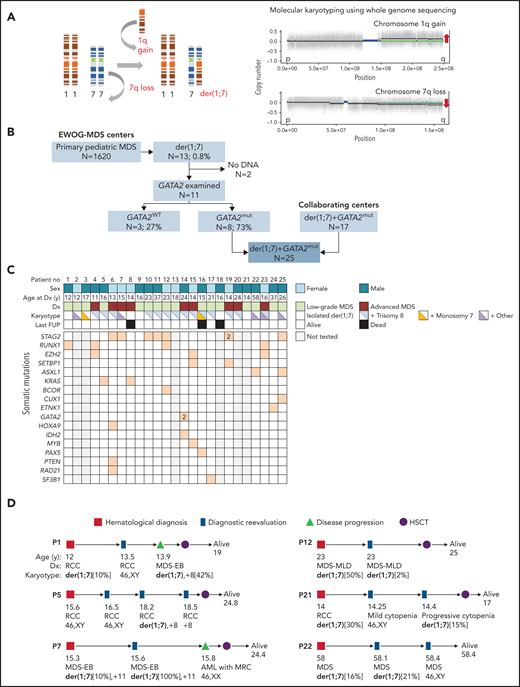
Association of chromosomal aberration der(1;7)(q10;p10) with GATA2 deficiency and MDS. (A) Left: schematic representation of the mechanism leading to the formation of unbalanced der(1;7) aberration that results in trisomy 1q and deletion 7q. Right: copy number variation profiling based on whole genome sequencing data of the patient P15 depicting gain of material of 1q and loss of material of 7q, indicated by red arrows. (B) Composition of the study cohort. Within the EWOG-MDS primary MDS cohort, 13 cases with the der(1;7) were encountered, 8 of which had germline GATA2 mutations. An additional 17 cases with GATA2-related MDS and der(1;7) were enrolled from collaborating institutions for a more comprehensive analysis. (C) Matrix plot depicting clinical and genetic data of GATA2-MDS patients with der(1;7). (D) Dynamics of der(1;7) clone. Spontaneous regression or loss of the der(1;7) as well as phenotype changes over the course of disease are shown. AML with MRC, acute myeloid leukemia with myelodysplasia-related changes; Dx, diagnosis; EWOG-MDS, European Working Group of MDS in Childhood; last FUP, last follow-up; MDS-EB, MDS with excess blasts; MDS-MLD, MDS with multilineage dysplasia; mut, mutated; no, number; RCC, refractory cytopenia of childhood; WT, wild type.
Association of chromosomal aberration der(1;7)(q10;p10) with GATA2 deficiency and MDS. (A) Left: schematic representation of the mechanism leading to the formation of unbalanced der(1;7) aberration that results in trisomy 1q and deletion 7q. Right: copy number variation profiling based on whole genome sequencing data of the patient P15 depicting gain of material of 1q and loss of material of 7q, indicated by red arrows. (B) Composition of the study cohort. Within the EWOG-MDS primary MDS cohort, 13 cases with the der(1;7) were encountered, 8 of which had germline GATA2 mutations. An additional 17 cases with GATA2-related MDS and der(1;7) were enrolled from collaborating institutions for a more comprehensive analysis. (C) Matrix plot depicting clinical and genetic data of GATA2-MDS patients with der(1;7). (D) Dynamics of der(1;7) clone. Spontaneous regression or loss of the der(1;7) as well as phenotype changes over the course of disease are shown. AML with MRC, acute myeloid leukemia with myelodysplasia-related changes; Dx, diagnosis; EWOG-MDS, European Working Group of MDS in Childhood; last FUP, last follow-up; MDS-EB, MDS with excess blasts; MDS-MLD, MDS with multilineage dysplasia; mut, mutated; no, number; RCC, refractory cytopenia of childhood; WT, wild type.
Expanding on the GATA2mut-der(1;7) association, we aimed to characterize this unique MDS subgroup in detail. We included 17 additional cases from collaborating institutions, resulting in a cohort of 25 patients with GATA2mut and der(1;7) (Figure 1B). This cohort consisted of children, adolescents, and adults with MDS arising from germline (constitutional) GATA2mut. Among the 25 patients, 26 germline GATA2mut were detected, of which 7 were novel (Table 1; supplemental Table 1). The germline mutational spectrum was comparable to what has been reported previously in the literature.18 Almost half of our cohort (12/25) had null alleles (8 frameshift/truncating, 3 nonsense, 1 splice site), 36% (9/25) carried missense mutations, and 16% (4/25) had variants in the intron 4 EBOX-GATA-ETS region (Table 1). We made several noteworthy genetic observations. Patient P15 carried 2 germline variants: a pathogenic truncating GATA2mut in addition to a missense variant of uncertain significance (p.T117I) not previously reported in the literature or ClinVar/gnomAD databases. Because a synonymous alteration affecting the same residue (p.T117T) was shown in multiple patients to cause missplicing and GATA2 RNA loss,19-21 we performed RNA-sequencing analysis from this patient’s marrow sample but did not identify an altered splice pattern arising from p.T117I alteration. Nonetheless, this variant might potentially affect protein function and act as a germline modifier. Double GATA2 germline mutations (cis p.T358N/p.L359V) have been previously described in 1 family.22 Of note, p.T358N was shown to have a loss-of-function and p.L359V a gain-of-function effect.23 Three GATA2 variants were identified in P14 in marrow, and fibroblast sequencing confirmed 1 as germline (p.W116LfsX69) and 2 as somatic (p.K390del and p.L305F). Whether the 2 somatic mutations arose in the same or different clones, and whether the somatic mutations occurred in cis or trans to modify the effect of the germline mutation is unknown with the available bulk sequencing data. Somatic GATA2mut are exceptionally rare in GATA2 deficiency. The p.K390del somatic mutation reported here was previously found in 1 patient with a germline GATA2mut and der(1;7); however, its functional consequence was not assessed.12 A recent study reported 1 case with somatic rescue mosaicism causing phenotypic reversion of the germline GATA2mut in hematopoiesis.24
Characteristics of patients with germline GATA2 mutations and der(1;7)(q10;p10)
| Parameter | |
|---|---|
| No. of patients | 25 |
| Age at diagnosis, y (median [range]) | 16 (11-58) |
| Sex, % (cases) | |
| Female | 40 (10/25) |
| Male | 60 (15/25) |
| Immunodeficiency | 48% (12/25) |
| Myeloid malignancy | |
| Low-grade MDS* | 64% (16/25) |
| Advanced MDS† | 36% (9/25) |
| GATA2 mutations, % (cases) | |
| Null (nonsense, frameshift, splice site | 48 (12/25) |
| Missense | 36 (9/25) |
| Intron 4 | 16 (4/25) |
| Cytogenetics, % (cases) | |
| der(1;7) only | 24 (6/25) |
| der(1;7) 1 trisomy 8‡ | 52 (13/25) |
| der(1;7) 1 monosomy 7 | 4 (1/25) |
| der(1;7) 1 trisomy 8 1 monosomy 7 | 4 (1/25) |
| der(1;7) 1 other aberrations | 16 (4/25) |
| Disappearing der(1;7) clone§ | 95% (18/19) |
| Somatic mutations | 24% (6/25) |
| Alive at last follow-up | 88% (22/25) |
| Parameter | |
|---|---|
| No. of patients | 25 |
| Age at diagnosis, y (median [range]) | 16 (11-58) |
| Sex, % (cases) | |
| Female | 40 (10/25) |
| Male | 60 (15/25) |
| Immunodeficiency | 48% (12/25) |
| Myeloid malignancy | |
| Low-grade MDS* | 64% (16/25) |
| Advanced MDS† | 36% (9/25) |
| GATA2 mutations, % (cases) | |
| Null (nonsense, frameshift, splice site | 48 (12/25) |
| Missense | 36 (9/25) |
| Intron 4 | 16 (4/25) |
| Cytogenetics, % (cases) | |
| der(1;7) only | 24 (6/25) |
| der(1;7) 1 trisomy 8‡ | 52 (13/25) |
| der(1;7) 1 monosomy 7 | 4 (1/25) |
| der(1;7) 1 trisomy 8 1 monosomy 7 | 4 (1/25) |
| der(1;7) 1 other aberrations | 16 (4/25) |
| Disappearing der(1;7) clone§ | 95% (18/19) |
| Somatic mutations | 24% (6/25) |
| Alive at last follow-up | 88% (22/25) |
Low-grade MDS includes MDS with no blast increase (peripheral blood <1%, bone marrow <5%). This includes refractory cytopenia of childhood (RCC).
Includes 1 patient each with 20% and 25% bone marrow blasts.
P23 carries a hyperdiploid clone with trisomies of chromosomes 14, 19, and 21 in addition to trisomy 8.
Five patients had no detectable clone at follow-up examination and one had significant reduction in clone size (Figure 1D).
The main clinical and genetic characteristics of the patients are summarized in supplemental Table 1 and depicted in Figure 1C. Median age at diagnosis was 16 years (range, 11-58 years) and clinical manifestations ranged from recurrent infections, cellular deficiencies, immune dysfunction to MDS. Among pediatric patients, 8 were diagnosed with low-grade MDS (refractory cytopenia of childhood, with <5% marrow blasts) and 7 with advanced disease (Table 1; supplemental Table 1; Figure 1C). Marrow examination of adult patients revealed low-grade MDS in 8 and advanced MDS in 2. Somatic mutations in myeloid malignancy genes were identified in 95% (18/19) of tested patients (supplemental Table 1; Figure 1C). STAG2, RUNX1, EZH2, SETBP1, ASXL1, and KRAS genes were recurrently mutated, whereas BCOR, CUX1, ETNK1, GATA2, HOXA9, IDH2, MYB, PAX5, PTEN, RAD21, and SF3B1 mutations were present in single cases. Many of these genes are known to be affected in GATA2 deficiency, including SETBP1, ASXL1, STAG2, RUNX1, and RAS pathway genes.12,25 Among adults with MDS and der(1;7) recurrent ASXL1, RUNX1, and EZH2 mutations were also described, with RUNX1 affecting the der(1;7) subgroup more often compared with monosomy 7 or del(7q).17 The presence of der(1;7) as a single denominator in patients harboring somatic mutations in various genes might suggest that the der(1;7) lesion is first to evolve, followed by acquisition of secondary mutations. We observed der(1;7) and somatic mutations at the same time point in 15/17 tested cases. In 2 patients, the detection of small somatic clones (P5: KRAS 3% VAF and P17: SF3B1 3% VAF) preceded the detection of der(1;7), it is possible, however, that der(1;7) was initially undetectable because of the small clone size. Given the strong rationale for early detection of MDS in GATA2 deficiency, we can advocate for a surveillance using routine metaphase karyotyping and fluorescence in situ hybridization to detect chromosomal aberrations including der(1;7), and additional next-generation sequencing panel testing to find leukemia driver mutations.
Hematopoietic stem cell transplantation (HSCT) was performed in 22 cases; 21 were alive at a median follow-up of 2 years after HSCT. Of the 3 patients who were not transplanted, 2 died during chemotherapy and 1 was alive with a short follow-up from diagnosis. Der(1;7) was detected initially in 20 and at a later time point in 5 patients (supplemental Table 1). We observed that der(1;7) can be accompanied by other cytogenetic lesions that affect the same clone or develop independently; co-occurring or independent trisomy 8 did arise in 56% (14/25) of patients, whereas independent monosomy 7 clones were encountered in 8% (2/25) (Table 1; supplemental Table 1). Serial cytogenetic studies revealed marked variability of der(1;7) clonal burden in 6 cases. During follow-up examinations, the clone disappeared in 5 of them (with later recurrence in 2) and regressed from 50% to 2% in 1 (Figure 1D). Of note, 1 of the patients (P7) with a transient der(1;7) had normal cytogenetics during progression to acute myeloid leukemia, suggesting that the der(1;7) MDS clone might have disappeared after being outcompeted by acute myeloid leukemia cells with normal karyotype, or it might have failed to grow and remain under the detection limit. Summarizing, we observed fluctuations in the der(1;7) clone size, with transient or complete disappearance of the aberration in one-quarter (6/25) of the cohort. However, this did not result in clinical remission, unlike reported in SAMD9L syndrome where monosomy 7 can be replaced by rescuing uniparental isodisomy 7q.26
The cellular consequences of der(1;7) are elusive. NUF2 (1q23.3), involved in chromosome segregation, was shown to be upregulated in der(1;7) cases.17 Another study evaluated expression and methylation levels in der(1;7) adult patients.27 Although 96% of altered 7q genes were downregulated, >50% of differentially expressed 1q genes were also downregulated despite trisomy 1q. This gene dosage discrepancy has been attributed to extensive hypermethylation detected at 1q. Notably, the authors also demonstrated hypermethylation in binding sites for hematopoietic transcription factors including GATA2 and RUNX1. It is possible that der(1;7) requires an interplay with GATA2 loss either at genetic or epigenetic levels. Further studies are warranted to understand why der(1;7) clones are selected in individuals with GATA2 deficiency.
In summary, our data indicate that der(1;7) is significantly overrepresented in GATA2mut in comparison with GATA2wt MDS in children and adolescents, placing it as a strong predictor for underlying GATA2 deficiency. The knowledge is relevant for diagnosis, surveillance, and faster therapy stratification. The co-occurrence of additional secondary mutations in leukemia driver genes (in 95% of evaluated cases) indicates a malignant phenotype that necessitates timely HSCT performed as upfront therapy in these patients. Decrease or disappearance of der(1;7) clone observed in one-quarter of patients was not associated with clinical improvement, and thus should not justify a deviation from HSCT, a well-established curative approach in GATA2 deficiency.
Acknowledgments
The authors thank all European Working Group of MDS in Childhood National Coordinators: Barbara De Moerloose (Belgium), Sophia Polychronopoulou (Greece), Krisztián Kállay (Hungary), Owen Smith (Ireland), Riccardo Masetti (Italy), Marek Ussowicz (Poland), Oksana Fabri (Slovakia), Albert Catala (Spain), Markus Schmugge (Switzerland); the Coordinating Study Center of the University Medical Center Freiburg, Germany (Maria Siskou-Zwecker, Alexandra Fischer, Wilfried Truckenmüller) for excellent project management; Dirk Lebrecht, Christina Jäger, Sandra Zolles, Sophie Krüger, Marco Teller, Ali-Riza Kaya, and Miriam Erlacher (Freiburg) for diagnostic work; and Elissa Furutani (Dana-Farber and Boston Children's Cancer and Blood Disorders Center, Boston, MA) for patients management and data collection.
The present study was supported by grants from ERA PerMed GATA2-HuMo by German Federal Ministry of Education and Research (BMBF) 2018-123/01KU1904, Fritz-Thyssen Foundation 10.17.1.026MN, Deutsche Krebshilfe Max-Eder-Nachwuchsgruppenprogramm 70109005, Deutsche Kinderkrebsstifung DKS 2017.03 (M.W.W.), José Carreras Leukemia Foundation (V.B.P.), Baden-Württemberg LGFG stipend (E.J.K.), BMBF MyPred Network 01GM1911A (M.W.W., G.G., C.M.N.), AIRC 5xmille MYNERVA (C.M.), Spanish Carlos III Health Institute INT20/00073, and Marató TV3 Foundation 202001-31 (J.C.), and NIDDK RC2 DK122533 (M.D.F.; A.S.).
Authorship
Contribution: E.J.K., A.Y., C.M.N., and M.W.W. designed the study and wrote the manuscript; V.B.P., S.H., S. Lei, S. Lewis, and M.W.W. performed genomic studies and data analysis; M.D., V.d.H., J.S., H.H., J.B., D.D.H., M.D.F., A.S., A.P.H., S.M.H., K.R.C., D.E.A., C.M., S.B.K., C.D.D., M.O., A.A.B., K.T., S.B., H.I., D.T., K.J., B. Strahm, P.N., A.Y., C.M.N., and M.W.W. were involved in the patient care, sample collection, testing, and reporting; G.G., B. Schlegelberger, E.J.K., H. Alaiz, H. Andrikovics, D.B., B.H.B., M.C., J.C., O.H., K.N.M., K.N., F.P., J.T., N.V.R., and Z.Z. analyzed cytogenetic data; and all authors contributed to the manuscript and provided final approval.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Marcin W. Wlodarski, St Jude Children’s Research Hospital, 262 Danny Thomas Pl, Memphis, TN 38103; e-mail: marcin.wlodarski@stjude.org.
The online version of this article contains a data supplement.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal