

Key Points
B-ALL progressing after CD19 CAR T-cell therapy carries poor prognosis and remains an unmet clinical need.
Blinatumomab, inotuzumab, and CAR T-cell retreatment can induce CR after post–CAR T progression, but remission duration and survival are limited.

Abstract
CD19-targeted chimeric antigen receptor (CAR) T-cell therapy has become a breakthrough treatment of patients with relapsed/refractory B-cell acute lymphoblastic leukemia (B-ALL). However, despite the high initial response rate, the majority of adult patients with B-ALL progress after CD19 CAR T-cell therapy. Data on the natural history, management, and outcome of adult B-ALL progressing after CD19 CAR T cells have not been described in detail. Herein, we report comprehensive data of 38 adult patients with B-ALL who progressed after CD19 CAR T therapy at our institution. The median time to progression after CAR T-cell therapy was 5.5 months. Median survival after post–CAR T progression was 7.5 months. A high disease burden at the time of CAR T-cell infusion was significantly associated with risk of post–CAR T progression. Thirty patients (79%) received salvage treatment of post–CAR T disease progression, and 13 patients (43%) achieved complete remission (CR), but remission duration was short. Notably, 7 (58.3%) of 12 patients achieved CR after blinatumomab and/or inotuzumab administered following post–CAR T failure. Multivariate analysis revealed that a longer remission duration from CAR T cells was associated with superior survival after progression following CAR T-cell therapy. In summary, overall prognosis of adult B-ALL patients progressing after CD19 CAR T cells was poor, although a subset of patients achieved sustained remissions to salvage treatments, including blinatumomab, inotuzumab, and reinfusion of CAR T cells. Novel therapeutic strategies are needed to reduce risk of progression after CAR T-cell therapy and improve outcomes of these patients.
Introduction
CD19-targeted chimeric antigen receptor (CAR) T-cell therapy has transformed the treatment landscape of relapsed/refractory (R/R) B-cell acute lymphoblastic leukemia (B-ALL) over the past several years. Several studies have shown excellent antileukemic activities with an exceptionally high complete remission (CR) rate,1-7 leading to US Food and Drug Administration approval of 1 CD19 CAR product, tisagenlecleucel, in the treatment of pediatric and young adults with R/R B-ALL. However, in adult patients with R/R B-ALL, the incidence of severe CAR T-cell–associated adverse events was significantly high. In addition, 10% to 30% of patients are refractory to CAR T cells, and 30% to 60% subsequently relapse after CD19 CAR T-cell therapy.1-8 Patients with ALL who were refractory to or relapsed after CD19 CAR T-cell therapy pose significant management challenges with limited data for prognosis and effective therapeutics.
Several groups have reported poor outcomes of patients with B-cell non-Hodgkin lymphoma and chronic lymphocytic leukemia progressing after CD19 CAR T-cell therapy.9-11 However, the detailed clinical outcomes and subsequent treatments of patients with B-ALL after CAR T-cell therapy have never been reported. With several ongoing, large, multicenter, and international CD19 CAR T-cell trials in adult ALL, along with commercially available CD19 and CD22 directed antibody-based immunotherapy, understanding natural courses of patients with post–CAR T progression will enable us to better navigate treatments for these complicated but increasing number of patients. We therefore performed a comprehensive analysis and described the details of clinical characteristics, post–CAR T treatments, and outcomes of adult patients with B-ALL who progressed after CD19-targeted CAR T-cell therapy who were treated at Memorial Sloan Kettering Cancer Center (MSKCC).
Methods
Patients
We previously reported results of a single-center, phase 1 trial of our CD19 (19-28z) CAR T cells for R/R adult B-ALL at MSKCC (#NCT01044069). Details of the 19-28z CAR T-cell manufacturing process and treatment protocols have been previously described.1,12,13
We reviewed electronic records of all consented patients and described the characteristics, including treatments before and after CAR T cells, response to post–CAR T-salvage therapy, and outcomes of patients who progressed after the CD19 CAR T-cell therapy. Systemic therapies for B-ALL, including as bridging therapy before CAR T-cell therapy or as salvage treatment of post–CAR T progression, were abstracted from the available medical records. Both the initial study protocol and this retrospective analysis were reviewed and approved by the MSKCC Institutional Review Board. The cutoff date for data analysis was 31 December 2019.
Assessments and outcomes
Bridging and post-CAR T-salvage therapies were classified as either intensive or nonintensive based on the treatment’s myelosuppressive potential. Intensive therapies included regimens containing multiagent chemotherapy with/without tyrosine kinase inhibitors. Nonintensive therapy included minimally myelosuppressive chemotherapy with/without tyrosine kinase inhibitors or noncellular immunotherapy such as blinatumomab and inotuzumab ozogamicin.
Chromosomal abnormalities were stratified according to the Medical Research Council United Kingdom Acute Lymphoblastic Leukemia XII/Eastern Cooperative Oncology Group E2993 classification.14
Cytokine release syndrome (CRS) and immune effector cell–associated neurotoxicity syndrome (ICANS) were graded according to the American Society of Transplant and Cellular Therapy Grading for CRS and ICANS.15
Complete remission (CR) was defined as bone marrow (BM) blast percentage <5% (irrespective of minimal residual disease [MRD] status) and resolution of extramedullary (EM) disease as assessed by radiographic studies. MRD was assessed in BM aspirate samples by using multiparameter flow cytometry with sensitivity of at least 10−4 of total leukocyte events. Relapse was defined as the reappearance of blasts in blood or BM (≥5% lymphoblasts) or in an EM site after achievement of CR. Refractory disease was defined as failure to attain morphologic CR (ie, BM lymphoblast <5% and resolution of EM diseases).
Event-free survival (EFS) was defined as the time from CAR T-cell infusion until confirmed refractory disease, morphologic relapse, or death; patients not known to have any of these events were censored on date of last follow-up. Overall survival (OS) of the entire cohort (N = 56) was defined as the time from CAR T-cell infusion to last contact or death, whichever occurred first. OS post–CAR T progression was assessed for a subset of patients who progressed (n = 38) and was defined as time from progression post–CAR T-cell therapy until last contact or death.
Statistical analysis
Continuous variables are described by using medians and range. Categorical variables are reported in numbers and percentage. EFS, OS, and OS post–CAR T progression were estimated by using the Kaplan-Meier method. Univariate and multivariate analyses for factors associated with survival were performed with the use of Cox models. Receipt of salvage chemotherapy and noncellular immunotherapy were evaluated as time-dependent covariates. Cumulative incidence of R/R disease was calculated, with death as a competing risk and compared between various covariates using cause-specific hazard models. All reported P values were 2-sided; values of P < .05 were considered statistically significant. Factors of P ≤ .10 from the univariate analysis were included in the multivariable model for OS post–CAR T progression. Analyses were performed by using R software, version 3.6.2 (R Foundation for Statistical Computing).
Results
Outcomes of patients treated with CD19 CAR T cells
Between May 2010 and March 2017, a total of 56 adult patients with B-ALL were treated with 19-28z CAR T cells at MSKCC (supplemental Table 1, available on the Blood Web site). Of the 56 treated patients, 54 (96%) were evaluable for response; 2 were not evaluable for response due to early treatment-associated mortality as previously reported.1 Forty-five patients (83.3%) achieved CR (35 with negative MRD; 8 with detectable MRD; and 2 with unknown MRD status), and 9 patients were refractory to the therapy. Severe CRS and ICANS were observed in 15 (27%) and 24 (43%) patients, respectively. Tocilizumab was given in 20 patients (36%), with a median time from infusion to administration of 7 days (3-17 days). Nineteen patients (34%) received corticosteroid. The median time from CAR T-cell infusion to corticosteroid was 8 days (3-15 days). With a median follow-up of 44.7 months (interquartile range, 20.8-65.4 months), 38 patients (68%) relapsed or failed to achieve morphologic CR after CAR T-cell therapy. All 8 patients who attained MRD+ CR after CAR T cells eventually developed morphologic relapse. The median OS of the entire cohort was 13 months (95% confidence interval [CI], 9.11-23.34). The 3-year EFS and OS were 16.1% (95% CI, 8.8-29) and 26.6% (95% CI, 17-42), respectively (Figure 1A-B). The 1-year cumulative incidence of R/R disease was 67.9% (95% CI, 53.6-78.6) (Figure 1C).
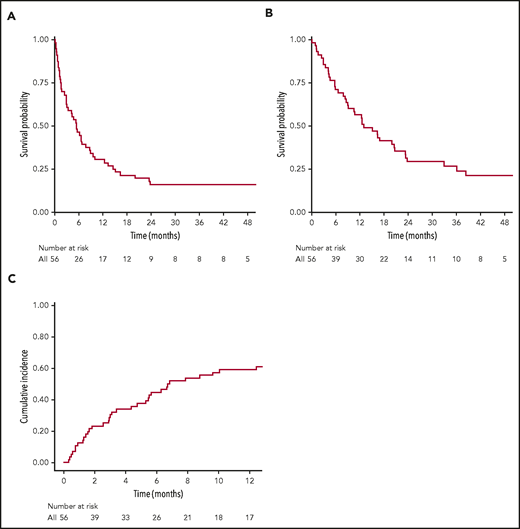
Survival outcomes and cumulative incidence of relapsed/refractory disease in 56 patients after CD19 CAR T-cell therapy. (A) EFS (3-year EFS, 16.1%; 95% CI, 8.8-29). (B) OS (3-year OS, 26.6%; 95% CI, 17-42). (C) Cumulative incidence of R/R disease (1-year cumulative incidence of R/R disease, 67.9%; 95% CI, 53.6-78.6).
Survival outcomes and cumulative incidence of relapsed/refractory disease in 56 patients after CD19 CAR T-cell therapy. (A) EFS (3-year EFS, 16.1%; 95% CI, 8.8-29). (B) OS (3-year OS, 26.6%; 95% CI, 17-42). (C) Cumulative incidence of R/R disease (1-year cumulative incidence of R/R disease, 67.9%; 95% CI, 53.6-78.6).
Factors associated with progression after CD19 CAR T-cell therapy
Table 1 summarizes the detailed characteristics of 38 patients who either failed to achieve CR (n = 9 [24%]) or who relapsed after CD19 CAR T-cell therapy (n = 29 [76%]). The median age was 45 years (23-74 years). Twenty-one patients (55%) had high-risk cytogenetic abnormalities, 17 patients (45%) had prior allogeneic hematopoietic stem cell transplantation (alloHSCT), and 17 patients (45%) had Philadelphia chromosome–positive ALL. The median number of treatments before CAR T-cell therapy was 3 lines (1-7 lines).
Baseline characteristics of 38 patients who progressed and 18 patients without disease progression after CAR T-cell therapy
| Characteristic | Relapse after CAR T-cells, n = 38 | No relapse after CAR T-cells, n = 18 | P |
|---|---|---|---|
| Median age at CD19 CAR T-cell therapy (range), y | 42.5 (23-74) | 55 (23-68) | .21 |
| Male sex | 28 (73.7%) | 14 (77.8%) | 1.00 |
| Median lines of treatment before CAR T-cell therapy (range) | 3 (2-7) | 2 (1-7) | .02 |
| Philadelphia chromosome–positive ALL (%) p190 p210 | 13 (34.2) 8 (21.1) 5 (13.1) | 4 (22.2) 3 (16.7) 1 (5.6) | .74 |
| Prior treatment with noncellular immunotherapy*(%) Blinatumomab Inotuzumab ozogamicin | 10 (26.3) 3 (7.9) | 3 (16.7) 1 (5.6) | 1.00 |
| Prior HSCT before CAR T-cell therapy (%) | 17 (44.7) | 3 (16.7) | .11 |
| Bridging therapy†(%) Intensive therapy Nonintensive therapy None | 10 (26.3) 21 (55.3) 7 (18.4) | 10 (55.6) 7 (38.9) 1 (5.6) | .12 |
| Disease burden before CAR T-cell therapy (%) Morphologic disease MRD No evidence of disease | 26 (68.4) 11 (28.9) 1 (2.6) | 5 (27.8) 6 (33.3) 6 (33.3) | .01 |
| CRS post-CAR T-cell therapy (%) Grade 3 or higher CRS‡ | 32 (84.2) 9 (23.7) | 16 (88.9) 6 (33.3) | .52 |
| ICANS (%) Grade 3 or higher ICANS | 22 (57.9) 14 (36.8) | 12 (66.7) 10 (55.6) | .19 |
| Tocilizumab for CRS/ICANS management (%) | 15 (39.5) | 5 (27.8) | .39 |
| Systemic corticosteroid for CRS/ICANS management (%) | 12 (31.6) | 7 (38.9) | .59 |
| Median time from CAR T-cell infusion to corticosteroid (range, d) | 7 (5-15) | 8 (3-12) | .80 |
| Median duration of systemic corticosteroid (range, d) | 7 (1-15) | 9 (2-27) | .16 |
| Median dose intensity of steroid (prednisone equivalent, mg/kg/d) | 1.37 (0.72-2.58) | 1.54 (0.79-6.75) | .38 |
| HSCT consolidation after CAR T-cell therapy (%) | 6 (15.8) | 9 (50.0) | .10 |
| Characteristic | Relapse after CAR T-cells, n = 38 | No relapse after CAR T-cells, n = 18 | P |
|---|---|---|---|
| Median age at CD19 CAR T-cell therapy (range), y | 42.5 (23-74) | 55 (23-68) | .21 |
| Male sex | 28 (73.7%) | 14 (77.8%) | 1.00 |
| Median lines of treatment before CAR T-cell therapy (range) | 3 (2-7) | 2 (1-7) | .02 |
| Philadelphia chromosome–positive ALL (%) p190 p210 | 13 (34.2) 8 (21.1) 5 (13.1) | 4 (22.2) 3 (16.7) 1 (5.6) | .74 |
| Prior treatment with noncellular immunotherapy*(%) Blinatumomab Inotuzumab ozogamicin | 10 (26.3) 3 (7.9) | 3 (16.7) 1 (5.6) | 1.00 |
| Prior HSCT before CAR T-cell therapy (%) | 17 (44.7) | 3 (16.7) | .11 |
| Bridging therapy†(%) Intensive therapy Nonintensive therapy None | 10 (26.3) 21 (55.3) 7 (18.4) | 10 (55.6) 7 (38.9) 1 (5.6) | .12 |
| Disease burden before CAR T-cell therapy (%) Morphologic disease MRD No evidence of disease | 26 (68.4) 11 (28.9) 1 (2.6) | 5 (27.8) 6 (33.3) 6 (33.3) | .01 |
| CRS post-CAR T-cell therapy (%) Grade 3 or higher CRS‡ | 32 (84.2) 9 (23.7) | 16 (88.9) 6 (33.3) | .52 |
| ICANS (%) Grade 3 or higher ICANS | 22 (57.9) 14 (36.8) | 12 (66.7) 10 (55.6) | .19 |
| Tocilizumab for CRS/ICANS management (%) | 15 (39.5) | 5 (27.8) | .39 |
| Systemic corticosteroid for CRS/ICANS management (%) | 12 (31.6) | 7 (38.9) | .59 |
| Median time from CAR T-cell infusion to corticosteroid (range, d) | 7 (5-15) | 8 (3-12) | .80 |
| Median duration of systemic corticosteroid (range, d) | 7 (1-15) | 9 (2-27) | .16 |
| Median dose intensity of steroid (prednisone equivalent, mg/kg/d) | 1.37 (0.72-2.58) | 1.54 (0.79-6.75) | .38 |
| HSCT consolidation after CAR T-cell therapy (%) | 6 (15.8) | 9 (50.0) | .10 |
One patient received both blinatumomab and inotuzumab ozogamicin before CD19 CAR T-cell therapy.
Intensive therapy included multiagent, myelosuppressive, chemotherapy-based regimen, and nonintensive regimen included nonmyelosuppressive chemotherapy or antibody-based therapy (eg, blinatumomab, inotuzumab). Breakdown of bridging intensity according to disease burden is provided in supplemental Figure 1.
Per American Society of Transplant and Cellular Therapy criteria.
Of the 38 patients progressing after CD19 CAR T-cell therapy, 36 (95%) had morphologic disease at the time of documented progression, whereas 2 (5%) initially relapsed with MRD+ disease (supplemental Table 2). Among the 29 patients who relapsed after initial response to the CAR T-cell therapy, the median time from CAR T-cell infusion to relapse was 5.5 months (1.3-24 months), and 6 of 29 patients had prior consolidative alloHSCT after achieving CR to CAR T-cell therapy. BM was the most common site of disease at the time of post–CAR T failure (n = 35), with a median 81% BM blasts at morphologic disease (8% to 100%). EM disease was observed in 13 patients (34.2%), with the central nervous system as the most common site of involvement (n = 4). Among patients with EM involvement, 3 had isolated EM disease (n = 1, bone; n = 1, central nervous system; n = 1, lymph node). Immunophenotypes at the time of progression were available in 30 patients (78.9%). CD19− disease was observed in 5 patients (16.7%), including 1 patient with the diagnosis of Philadelphia chromosome–positive acute mixed phenotypic leukemia (coexpressing myeloid and B lymphoid immunophenotypes) who relapsed with CD19− myeloid leukemia. There was a trend toward the longer median time from CAR T-cell therapy to CD19− relapse (6.8 months compared with 3.7 months for CD19+ relapse), but the difference was not statistically significant (P = .08).
Among clinical factors examined, a higher disease burden (BM blasts ≥5% or EM disease) before CAR T-cell infusion was the only risk factor significantly associated with the risk of post–CAR T progression (hazard ratio [HR], 2.2; 95% CI, 1.1-4.4; P = .02) (Table 2). Notably, previous exposure to blinatumomab or inotuzumab before CAR T-cell therapy was not associated with risk of progression after CAR T-cell therapy. Immune-mediated adverse events such as CRS and ICANS, severity of CRS/ICANS, tocilizumab, and systemic corticosteroid exposure, and post–CAR T alloHSCT consolidation did not affect the risk of progression after CAR T-cell therapy. Subgroup analysis was performed in patients with morphologic disease before CAR T-cell therapy and revealed no significant prognostic factor for progression post–CAR T in these patients (supplemental Table 3).
Univariate analysis for factors associated with risk of R/R disease after CD19 CAR T-cell therapy
| Univariate analysis | ||
|---|---|---|
| Parameter | HR (95% CI) | P |
| Age (>40 y vs ≤40 y) | 1.11 (0.58-2.10) | .8 |
| Sex (male vs female) | 0.75 (0.37-1.55) | .5 |
| Prior lines of treatment before CAR T cells (>3 vs ≤3) | 1.55 (0.81-2.95) | .2 |
| Philadelphia chromosome–positive ALL (yes vs no) | 1.06 (0.54-2.07) | .9 |
| Prior HSCT before CAR T cells (yes vs no) | 1.43 (0.75-2.71) | .3 |
| Prior blinatumomab before CAR T cells (yes vs no) | 1.09 (0.53-2.25) | .8 |
| Prior inotuzumab ozogamicin before CAR T cells (yes vs no) | 0.93 (0.28-3.05) | .9 |
| Bridging therapy before CAR T cells (no or nonintensive vs intensive therapy) | 1.32 (0.64-2.71) | .4 |
| Baseline LDH before lymphodepletion (normal vs high) | 0.93 (0.48-1.83) | .8 |
| Baseline platelet count before lymphodepletion (normal vs low) | 0.73 (0.37-1.43) | .4 |
| Disease status before lymphodepletion (morphologic vs MRD) | 2.20 (1.09-4.41) | .02 |
| Lymphodepletion chemotherapy (fludarabine-containing regimen vs cyclophosphamide) | 1.05 (0.67-1.65) | .8 |
| CRS (yes vs no) | 0.73 (0.30-1.75) | .5 |
| ICANS (yes vs no) | 0.63 (0.33-1.20) | .2 |
| Steroid exposure during CAR T-cell therapy (yes vs no) | 0.98 (0.49-1.95) | .9 |
| Tocilizumab exposure during CAR T-cell therapy (yes vs no) | 1.35 (0.70-2.60) | .4 |
| Duration of steroid exposure (d) | 0.95 (0.83-1.08) | .4 |
| Steroid dose intensity during CAR T-cell therapy (mg/kg/d) | 0.77 (0.20-3.03) | .7 |
| HSCT consolidation after CAR T-cell therapy | 0.55 (0.23-1.32) | .2 |
| Univariate analysis | ||
|---|---|---|
| Parameter | HR (95% CI) | P |
| Age (>40 y vs ≤40 y) | 1.11 (0.58-2.10) | .8 |
| Sex (male vs female) | 0.75 (0.37-1.55) | .5 |
| Prior lines of treatment before CAR T cells (>3 vs ≤3) | 1.55 (0.81-2.95) | .2 |
| Philadelphia chromosome–positive ALL (yes vs no) | 1.06 (0.54-2.07) | .9 |
| Prior HSCT before CAR T cells (yes vs no) | 1.43 (0.75-2.71) | .3 |
| Prior blinatumomab before CAR T cells (yes vs no) | 1.09 (0.53-2.25) | .8 |
| Prior inotuzumab ozogamicin before CAR T cells (yes vs no) | 0.93 (0.28-3.05) | .9 |
| Bridging therapy before CAR T cells (no or nonintensive vs intensive therapy) | 1.32 (0.64-2.71) | .4 |
| Baseline LDH before lymphodepletion (normal vs high) | 0.93 (0.48-1.83) | .8 |
| Baseline platelet count before lymphodepletion (normal vs low) | 0.73 (0.37-1.43) | .4 |
| Disease status before lymphodepletion (morphologic vs MRD) | 2.20 (1.09-4.41) | .02 |
| Lymphodepletion chemotherapy (fludarabine-containing regimen vs cyclophosphamide) | 1.05 (0.67-1.65) | .8 |
| CRS (yes vs no) | 0.73 (0.30-1.75) | .5 |
| ICANS (yes vs no) | 0.63 (0.33-1.20) | .2 |
| Steroid exposure during CAR T-cell therapy (yes vs no) | 0.98 (0.49-1.95) | .9 |
| Tocilizumab exposure during CAR T-cell therapy (yes vs no) | 1.35 (0.70-2.60) | .4 |
| Duration of steroid exposure (d) | 0.95 (0.83-1.08) | .4 |
| Steroid dose intensity during CAR T-cell therapy (mg/kg/d) | 0.77 (0.20-3.03) | .7 |
| HSCT consolidation after CAR T-cell therapy | 0.55 (0.23-1.32) | .2 |
LDH, lactate dehydrogenase.
Salvage treatments and outcomes of patients progressing after CD19 CAR T-cell therapy
To evaluate which salvage treatments are most effective for progression after CD19 CAR T-cell therapy, we examined the details of post–CAR T-salvage treatments. Figure 2 illustrates the treatment sequence among 38 patients progressing after CD19 CAR T-cell therapy. Systemic treatments were classified as intensive, nonintensive, and noncellular immunotherapy. Details of salvage therapy and treatment responses are summarized in supplemental Tables 4 to 6.

Distribution of treatment in patients who progressed after CAR T-cell therapy. Consort diagram illustrates the treatment sequence and distribution of salvage therapy in 38 patients who progressed after CD19 CAR T-cell therapy.
Distribution of treatment in patients who progressed after CAR T-cell therapy. Consort diagram illustrates the treatment sequence and distribution of salvage therapy in 38 patients who progressed after CD19 CAR T-cell therapy.
Among the 38 patients who progressed after CAR T cells, 30 (79%) received subsequent salvage treatments, and 8 (21%) received only palliative corticosteroid or supportive care. Of the 30 patients who received post–CAR T-salvage treatments, 6 (20%) received intensive multiagent chemotherapy; 18 (60%) received nonintensive therapy; 3 (10%) received either blinatumomab or inotuzumab; and 3 (10%) had reinfusion of CAR T cells as the first immediate salvage therapy following CD19 CAR T cells. Twenty-four patients (80%) were evaluable for response, and 6 patients were not evaluable due to early mortality or lost to follow-up. Of 24 evaluable patients, 8 (33.3%) achieved CR to the first salvage therapy. Nineteen patients (14 of 16 who had persistent disease and 5 of 8 who relapsed after first salvage therapy) subsequently received additional salvage treatments (Figure 2). Among patients who did not achieve CR with the first salvage therapies, 4 attained CR after the second line, and 1 achieved CR after ≥3 lines of salvage treatment. The overall median number of salvage therapy for progression after CAR T cells was 2 lines (1-6 lines). The median remission duration was 4.5 months (2.1-32.1 months), and the median EFS in patients who achieved CR to the salvage therapy was 5.8 months (4.1 months to months not available) (Figure 3A).
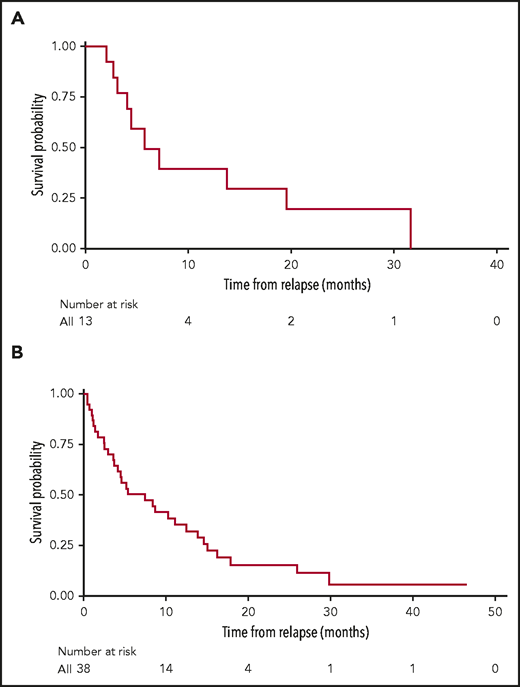
Survival outcomes in patients who progressed after CD19 CAR T-cell therapy. (A) EFS of patients who achieved CR to salvage treatments for post-CAR T disease progression. Median EFS was 4.5 months (2.1-32.1 months). (B) OS from post–CAR T progression of 38 patients who progressed after CD19 CAR T-cell therapy. Median OS was 7.5 months (95% CI, 4.2-13.9 months) with corresponding 1-year OS of 35% (95% CI, 23-56).
Survival outcomes in patients who progressed after CD19 CAR T-cell therapy. (A) EFS of patients who achieved CR to salvage treatments for post-CAR T disease progression. Median EFS was 4.5 months (2.1-32.1 months). (B) OS from post–CAR T progression of 38 patients who progressed after CD19 CAR T-cell therapy. Median OS was 7.5 months (95% CI, 4.2-13.9 months) with corresponding 1-year OS of 35% (95% CI, 23-56).
Seven patients (18%) underwent alloHSCT after salvage treatments for post–CAR T progression (first alloHSCT, n = 5; second alloHSCT, n = 2). Disease status at the time of transplant included 1 in MRD− CR, 5 in MRD+ CR, and 1 in CR with unknown MRD. The median time from post–CAR T progression to alloHSCT was 99 days (31-231 days). At the time of data cutoff, 4 patients relapsed after alloHSCT and died of progressive disease, 2 died of transplant-related mortality, and 1 has remained alive in continuous remission. Supplemental Table 7 summarizes the details of patients who underwent alloHSCT for post–CAR T progression.
Overall, 31 (82%) of 38 patients who progressed after CAR T-cell therapy died. Causes of death included disease progression (n = 24 [77%]), salvage treatment–related mortality (n = 3 [10%]), and unknown etiology due to inadequate documents or lost to follow-up (n = 4 [13%]). With the median follow-up of 20 months after post–CAR T progression, the median OS in 38 patients was 7.5 months (95% CI, 4.14-13.9 months) with a 1-year OS of 35% (95% CI, 23-56) (Figure 3B). We investigated factors associated with survival among the patients progressing after CD19 CAR T-cell therapy. In a univariate analysis, receipt of bridging therapy before CAR T-cell infusion was associated with a shorter OS after post–CAR T progression, with patients who received high-intensity bridging therapy having inferior OS, but this association was not observed in the multivariate analysis (Table 3). There was a nonsignificant trend toward improved OS (HR, 0.48; 95% CI, 0.17-1.35; P = .16) in patients who received salvage therapy for progression after CAR T cells. However, prior treatments with blinatumomab or inotuzumab, relapse site, CD19 expression, and BM blast percentage at the time of post–CAR T progression were not associated with mortality risk. Although we did not find that the patients who received post–CAR T-salvage therapy had significantly improved survival, patients who achieved CR from salvage therapy had a lower risk of mortality compared with patients who did not achieve CR (HR, 0.31; 95% CI, 0.11-0.87; P = .03). The median OS of patients who achieved CR to salvage therapy was 16.2 months (95% CI, 8.4 months to months not available), whereas patients who did not respond to salvage treatment had a median OS of 4.6 months (95% CI, 3.6-13.9). We performed a multivariate analysis with factors with values of P ≤. 1 from univariate Cox regression analysis and found time from CAR T-cell therapy to progression was the only independent prognostic factor of survival following post–CAR T progression (HR, 0.91; 95% CI, 0.83-1.00; P = .04).
Univariate and multivariate analysis of factors associated with survival following progression after CD19 CAR T-cell therapy
| Univariate analysis | Multivariate analysis | |||
|---|---|---|---|---|
| Parameter | HR (95% CI) | P | HR (95% CI) | P |
| Age (>40 vs ≤40 y) | 1.64 (0.80-3.34) | .20 | — | — |
| Sex (male vs female) | 0.53 (0.21-1.34) | .20 | — | — |
| Philadelphia chromosome–positive ALL (yes vs no) | 0.63 (0.29-1.35) | .20 | — | — |
| History of prior HSCT before CAR T cells (yes vs no) | 0.63 (0.30-1.30) | .20 | — | — |
| Prior blinatumomab or inotuzumab before CAR T cells (yes vs no) | 1.78 (0.79-4.0) | .20 | — | — |
| Bridging therapy before CAR T-cell infusion (no or nonintensive vs intensive) | 0.36 (0.16-0.83) | .02 | 0.41 (0.14-1.16) | .09 |
| Time from CAR T-cell infusion to relapse in months | 0.94 (0.86-1.02) | .09 | 0.91 (0.83-1.00) | .04 |
| BM blasts at the time of relapse | 1.01 (1.00-1.03) | .08 | 1.01 (0.99-1.03) | .2 |
| Extramedullary relapse (yes vs no) | 0.85 (0.4-1.82) | .70 | — | — |
| CD19 expression at relapse (positive vs negative) | 0.63 (0.22-1.76) | .40 | — | — |
| Salvage therapy after relapse (yes vs no) | 0.48 (0.17-1.35) | .16 | — | — |
| Blinatumomab or inotuzumab as salvage therapy after relapse (yes vs no) | 1.41 (0.60-3.33) | .43 | — | — |
| Univariate analysis | Multivariate analysis | |||
|---|---|---|---|---|
| Parameter | HR (95% CI) | P | HR (95% CI) | P |
| Age (>40 vs ≤40 y) | 1.64 (0.80-3.34) | .20 | — | — |
| Sex (male vs female) | 0.53 (0.21-1.34) | .20 | — | — |
| Philadelphia chromosome–positive ALL (yes vs no) | 0.63 (0.29-1.35) | .20 | — | — |
| History of prior HSCT before CAR T cells (yes vs no) | 0.63 (0.30-1.30) | .20 | — | — |
| Prior blinatumomab or inotuzumab before CAR T cells (yes vs no) | 1.78 (0.79-4.0) | .20 | — | — |
| Bridging therapy before CAR T-cell infusion (no or nonintensive vs intensive) | 0.36 (0.16-0.83) | .02 | 0.41 (0.14-1.16) | .09 |
| Time from CAR T-cell infusion to relapse in months | 0.94 (0.86-1.02) | .09 | 0.91 (0.83-1.00) | .04 |
| BM blasts at the time of relapse | 1.01 (1.00-1.03) | .08 | 1.01 (0.99-1.03) | .2 |
| Extramedullary relapse (yes vs no) | 0.85 (0.4-1.82) | .70 | — | — |
| CD19 expression at relapse (positive vs negative) | 0.63 (0.22-1.76) | .40 | — | — |
| Salvage therapy after relapse (yes vs no) | 0.48 (0.17-1.35) | .16 | — | — |
| Blinatumomab or inotuzumab as salvage therapy after relapse (yes vs no) | 1.41 (0.60-3.33) | .43 | — | — |
Factors with values of P ≤ .1 in univariate analysis were included in multivariate analysis.
Blinatumomab or inotuzumab in patients progressing after CD19 CAR T-cell therapy
To evaluate the role of CD19 and CD22-targeted noncellular immunotherapy in patients who progressed after CD19 CAR T-cell therapy, we examined the characteristics of patients who received either blinatumomab or inotuzumab after CD19 CAR T cells (Table 4). Twelve patients (40%) received either blinatumomab (n = 4), inotuzumab (n = 5), or both (n = 3) post–CAR T-cell therapy. Among these 12 patients, 3 patients (25%) received blinatumomab, and none received inotuzumab before CAR T-cell therapy.
Characteristics and outcomes of 12 patients who received blinatumomab and/or inotuzumab ozogamicin as a salvage therapy for post–CAR T disease progression
| Blinatumomab | Inotuzumab | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Drugs | Prior immuno-therapy exposure before CAR T-cell therapy | CD19 status at relapse after CAR T-cell therapy | Time from CAR T-cells to blinatumomab (d) | CRS (grade) | Neuro-toxicities (grade) | Cytopenia grade 3 or higher | Response to blinatumomab | Time from CAR T- cells to inotuzumab (d) | Cytopenia grade 3 or higher | SOS | Response to inotuzumab | Survival status at last contact | Cause of death |
| MSK15 | Blinatumomab | No | Positive | 1500 | Yes (grade 2) | Yes (grade 1) | No | MRD− CR | — | — | — | — | Alive | — |
| MSK34 | Blinatumomab | No | Positive | 318 | Yes (unknown) | Yes (unknown) | Unknown | MRD− CR | — | — | — | — | Dead | Disease |
| MSK39 | Blinatumomab | No | Positive | 54 | Yes (unknown) | Yes (unknown) | Unknown | CR | — | — | — | — | Alive | — |
| MSK56 | Blinatumomab | No | Positive | 361 | No | No | No | NR | — | — | — | — | Dead | Unknown |
| MSK26 | Inotuzumab | No | Negative | — | — | — | — | — | 253 | Grade 4 WBC; grade 4 platelet | Yes | Unknown | Dead | TRM |
| MSK44 | Inotuzumab | Blinatumomab | Negative | — | — | — | — | — | 564 | Grade 4 WBC; grade 4 platelet | No | MRDȒ CR | Alive | — |
| MSK46 | Inotuzumab | No | — | — | — | — | — | — | 484 | Grade 4 WBC | No | Unknown | Dead | Disease |
| MSK50 | Inotuzumab | Blinatumomab | Negative | — | — | — | — | — | 136 | Grade 4 WBC; grade 4 platelet | Yes (after alloHSCT) | MRD+ CR | Dead | Disease |
| MSK53 | Inotuzumab | Blinatumomab | Positive | — | — | — | — | — | 198 | Grade 4 WBC; grade 4 platelet | No | CR | Dead | Disease |
| MSK29 | Blinatumomab then inotuzumab | No | Positive | 895 | Yes (grade 1) | No | No | NR | 965 | Grade 3 WBC; grade 4 platelet | No | NR | Dead | Disease |
| MSK35 | Blinatumomab then inotuzumab | No | Positive | 73 | No | No | Unknown | NR | 116 | Grade 4 WBC; grade 4 platelet | No | MRD+ CR | Dead | TRM |
| MSK55 | Blinatumomab then inotuzumab | No | Positive | 90 | Yes (grade 1) | No | No | NR | 229 | Grade 4 WBC; grade 4 platelet | No | NR | Dead | Disease |
| Blinatumomab | Inotuzumab | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Drugs | Prior immuno-therapy exposure before CAR T-cell therapy | CD19 status at relapse after CAR T-cell therapy | Time from CAR T-cells to blinatumomab (d) | CRS (grade) | Neuro-toxicities (grade) | Cytopenia grade 3 or higher | Response to blinatumomab | Time from CAR T- cells to inotuzumab (d) | Cytopenia grade 3 or higher | SOS | Response to inotuzumab | Survival status at last contact | Cause of death |
| MSK15 | Blinatumomab | No | Positive | 1500 | Yes (grade 2) | Yes (grade 1) | No | MRD− CR | — | — | — | — | Alive | — |
| MSK34 | Blinatumomab | No | Positive | 318 | Yes (unknown) | Yes (unknown) | Unknown | MRD− CR | — | — | — | — | Dead | Disease |
| MSK39 | Blinatumomab | No | Positive | 54 | Yes (unknown) | Yes (unknown) | Unknown | CR | — | — | — | — | Alive | — |
| MSK56 | Blinatumomab | No | Positive | 361 | No | No | No | NR | — | — | — | — | Dead | Unknown |
| MSK26 | Inotuzumab | No | Negative | — | — | — | — | — | 253 | Grade 4 WBC; grade 4 platelet | Yes | Unknown | Dead | TRM |
| MSK44 | Inotuzumab | Blinatumomab | Negative | — | — | — | — | — | 564 | Grade 4 WBC; grade 4 platelet | No | MRDȒ CR | Alive | — |
| MSK46 | Inotuzumab | No | — | — | — | — | — | — | 484 | Grade 4 WBC | No | Unknown | Dead | Disease |
| MSK50 | Inotuzumab | Blinatumomab | Negative | — | — | — | — | — | 136 | Grade 4 WBC; grade 4 platelet | Yes (after alloHSCT) | MRD+ CR | Dead | Disease |
| MSK53 | Inotuzumab | Blinatumomab | Positive | — | — | — | — | — | 198 | Grade 4 WBC; grade 4 platelet | No | CR | Dead | Disease |
| MSK29 | Blinatumomab then inotuzumab | No | Positive | 895 | Yes (grade 1) | No | No | NR | 965 | Grade 3 WBC; grade 4 platelet | No | NR | Dead | Disease |
| MSK35 | Blinatumomab then inotuzumab | No | Positive | 73 | No | No | Unknown | NR | 116 | Grade 4 WBC; grade 4 platelet | No | MRD+ CR | Dead | TRM |
| MSK55 | Blinatumomab then inotuzumab | No | Positive | 90 | Yes (grade 1) | No | No | NR | 229 | Grade 4 WBC; grade 4 platelet | No | NR | Dead | Disease |
NR, no response; TRM, treatment-related mortality; WBC, white blood cell.
Blinatumomab (n = 7) was used as a first (n = 1), second (n = 4), and third (n = 2) line of post–CAR T-salvage therapy. At the time of blinatumomab initiation, 6 patients had evidence of morphologic disease, and 3 of these patients attained CR after blinatumomab (2 MRD−, 1 unknown MRD status). The patient who had MRD+ at blinatumomab initiation subsequently progressed with EM disease. The median time from CD19 CAR T-cell therapy to blinatumomab was 318 days (54-1500 days). No patient developed grade 3 or higher CRS/ICANS or other nonhematologic toxicities after blinatumomab.
Inotuzumab was used as salvage therapy for post–CAR T progression in 8 patients as a first (n = 2), second (n = 1), and third or beyond (n = 5) line of treatment. At the time of inotuzumab, all patients had morphologic disease, including 3 patients with CD19− disease. Four patients achieved CR (2 MRD+, 1 MRD−, 1 unavailable MRD status). Two patients developed severe adverse events of grade 4 infection with multiorgan dysfunction (n = 1) and grade 5 sinusoidal obstruction syndrome (SOS) (n = 1). Hepatic SOS was observed in 2 patients; the first patient had prior alloHSCT and developed grade 5 SOS during inotuzumab, and the second patient developed grade 4 SOS post-alloHSCT.
Among 7 patients who achieved remission after post-CAR T blinatumomab or inotuzumab, 2 underwent alloHSCT; 1 patient eventually relapsed and died of active disease, and the other patient died of transplant-related toxicities in remission. One patient who had pre-CAR T alloHSCT received donor lymphocyte infusion after achieving CR from blinatumomab and remained alive in CR at the time of last follow-up. Among 4 patients who did not undergo transplant after attaining CR from blinatumomab or inotuzumab salvage treatment, 2 relapsed and died of progressive disease, and the other 2 were lost to follow-up.
Retreatment with CD19 CAR T cells
Among patients who progressed after the initial CAR T-cell therapy, 10 (26.3%) subsequently received reinfusion of 19-28z CAR T cells for CD19+ disease. The 19-28z CAR T cells used for reinfusion were from the stored manufactured product left over from the initial treatment. Two patients received CAR T-cell retreatment for MRD detection, and 8 patients for morphologic disease. The median time from the initial CAR T-cell infusion to reinfusion was 185 days (80-1110 days). CAR T-cell reinfusion was used as a first (n = 3), second (n = 4), and third (n = 3) line of salvage therapy for post–CAR T progression. Data on patients who received CAR T-cell reinfusion are summarized in Table 5.
Characteristics and outcomes of 10 patients who received CD19 CAR T-cell retreatment of post–CAR T disease progression
| First CAR T-cell therapy | Second CAR T-cell therapy | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Disease status at first CAR T-cell therapy | Lympho- depletion | Dose (cells per kg) | Peak expansion (vcn/mL) | CRS maximum grade | ICANS maximum grade | Response to first CAR T-cell therapy | Relapse site/burden | Time from first CAR T to second CAR T (d) | Disease status at second CAR T-cell therapy | Lympho- depletion | Dose (/kg) | Peak CAR T expansion (vcn/mL) | CRS maximum grade | ICANS maximum grade | Response to second CAR T-cell therapy | Post-second CAR T treatment | Living status last contact |
| MSK7 | Morphologic | Cy 3 g/m2 | 3×106 | 62 719 | 1 | 3 | MRD− CR | BM/gross | 166 | Morphologic | Cy 1.5 g/m2 | 3×106 | 13 353 | 1 | No | NR | No | Dead (disease) |
| MSK9 | MRD | Cy 1.5 g/m2 | 0.4×106 | 5208 | No | No | MRD− CR | BM/MRD | 204 | MRD | Cy 3 g/m2 | 3×106 | 0 | 1 | No | NR | Yes | Dead (disease) |
| MSK13 | Morphologic | Cy 3 g/m2 | 3×106 | 2 320 302 | 3 | 3 | MRD− CR | BM/gross | 1041 | Morphologic | Flu/Cy | 1×106 | 746 | No | No | NR | Yes | Dead (disease) |
| MSK16 | MRD | Cy 3 g/m2 | 3×106 | 0 | 2 | 2 | MRD− CR | EM, BM/gross | 104 | MRD | Cy 3 g/m2 | 3×106 | 0 | No | No | MRD− CR | No | Dead (disease) |
| MSK17 | Morphologic | Cy 3 g/m2 | 3×106 | 197 262 | 4 | 4 | MRD− CR | BM/gross | 104 | Morphologic | Cy 2 g/m2 | 3×106 | 200 363 | 3 | 5 | MRD− CR | No | Dead (TRM) |
| MSK24 | Morphologic | Cy 3 g/m2 | 1×106 | 34 183 | 1 | No | MRD+ CR | BM/gross | 80 | Morphologic | Cy 3 g/m2 | 1×106 | 0 | 1 | No | NR | Yes | Dead (unknown) |
| MSK29 | MRD− | Cy 3 g/m2 | 3×106 | 26 210 | 1 | 2 | MRD− CR | BM/gross | 1110 | Morphologic | Flu/Cy | 3×106 | 35 800 | 1 | 1 | NR | No | Dead (unknown) |
| MSK36 | MRD | Cy 3 g/m2 | 3×106 | 0 | No | No | MRD+ CR | BM/gross | 343 | Morphologic | Flu/Cy | 1×106 | 177 758 | 1 | No | NR | Yes | Alive |
| MSK44 | Morphologic | Flu/Cy | 1×106 | 129 980 | 1 | 3 | MRD+ CR | EM, BM/gross | 451 | Morphologic | Cy 3 g/m2 | 1×106 | 0 | 2 | No | MRD+ CR | Yes | Alive |
| MSK56 | Morphologic | Flu/Cy | 1×106 | 20 400 | 2 | 0 | MRD+ CR | BM/MRD | 164 | MRD | Flu/Cy | 1×106 | 18 900 | 2 | No | MRD− CR | Yes | Dead (unknown) |
| First CAR T-cell therapy | Second CAR T-cell therapy | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Disease status at first CAR T-cell therapy | Lympho- depletion | Dose (cells per kg) | Peak expansion (vcn/mL) | CRS maximum grade | ICANS maximum grade | Response to first CAR T-cell therapy | Relapse site/burden | Time from first CAR T to second CAR T (d) | Disease status at second CAR T-cell therapy | Lympho- depletion | Dose (/kg) | Peak CAR T expansion (vcn/mL) | CRS maximum grade | ICANS maximum grade | Response to second CAR T-cell therapy | Post-second CAR T treatment | Living status last contact |
| MSK7 | Morphologic | Cy 3 g/m2 | 3×106 | 62 719 | 1 | 3 | MRD− CR | BM/gross | 166 | Morphologic | Cy 1.5 g/m2 | 3×106 | 13 353 | 1 | No | NR | No | Dead (disease) |
| MSK9 | MRD | Cy 1.5 g/m2 | 0.4×106 | 5208 | No | No | MRD− CR | BM/MRD | 204 | MRD | Cy 3 g/m2 | 3×106 | 0 | 1 | No | NR | Yes | Dead (disease) |
| MSK13 | Morphologic | Cy 3 g/m2 | 3×106 | 2 320 302 | 3 | 3 | MRD− CR | BM/gross | 1041 | Morphologic | Flu/Cy | 1×106 | 746 | No | No | NR | Yes | Dead (disease) |
| MSK16 | MRD | Cy 3 g/m2 | 3×106 | 0 | 2 | 2 | MRD− CR | EM, BM/gross | 104 | MRD | Cy 3 g/m2 | 3×106 | 0 | No | No | MRD− CR | No | Dead (disease) |
| MSK17 | Morphologic | Cy 3 g/m2 | 3×106 | 197 262 | 4 | 4 | MRD− CR | BM/gross | 104 | Morphologic | Cy 2 g/m2 | 3×106 | 200 363 | 3 | 5 | MRD− CR | No | Dead (TRM) |
| MSK24 | Morphologic | Cy 3 g/m2 | 1×106 | 34 183 | 1 | No | MRD+ CR | BM/gross | 80 | Morphologic | Cy 3 g/m2 | 1×106 | 0 | 1 | No | NR | Yes | Dead (unknown) |
| MSK29 | MRD− | Cy 3 g/m2 | 3×106 | 26 210 | 1 | 2 | MRD− CR | BM/gross | 1110 | Morphologic | Flu/Cy | 3×106 | 35 800 | 1 | 1 | NR | No | Dead (unknown) |
| MSK36 | MRD | Cy 3 g/m2 | 3×106 | 0 | No | No | MRD+ CR | BM/gross | 343 | Morphologic | Flu/Cy | 1×106 | 177 758 | 1 | No | NR | Yes | Alive |
| MSK44 | Morphologic | Flu/Cy | 1×106 | 129 980 | 1 | 3 | MRD+ CR | EM, BM/gross | 451 | Morphologic | Cy 3 g/m2 | 1×106 | 0 | 2 | No | MRD+ CR | Yes | Alive |
| MSK56 | Morphologic | Flu/Cy | 1×106 | 20 400 | 2 | 0 | MRD+ CR | BM/MRD | 164 | MRD | Flu/Cy | 1×106 | 18 900 | 2 | No | MRD− CR | Yes | Dead (unknown) |
Cy, cyclophosphamide; Flu, fludarabine; NR, no response; TRM, treatment-related mortality.
CRS of any grade was observed in 8 patients (80%), mostly grade 1 (n = 5) and grade 2 (n = 2). One patient experienced grade 3 CRS and received tocilizumab. The median time to CRS onset was 2 days (0-4 days) and lasted for a median duration of 4 days (1-9 days). ICANS was observed in 2 patients (grade 1, n = 1; grade 5, n = 1) with onset of 1 and 7 days after CAR T-cell infusion, respectively. The patient who developed grade 5 ICANS previously experienced grade 4 CRS and grade 4 ICANS with the initial CAR T-cell infusion 3 months earlier, but had completely recovered from these symptoms at the time of reinfusion.
Four patients received fludarabine and cyclophosphamide, and 6 patients received cyclophosphamide conditioning chemotherapy before reinfusion of CAR T cells (Table 5). Four patients (40%) were in CR after reinfusion of CAR T cells (2 with MRD− disease). Two of 3 patients who received the CAR T-cell reinfusion as the first immediate salvage treatment achieved CR (median duration from the first to second CAR T-cell infusion, 185 days), whereas 2 of 7 patients who received the CAR T-cell reinfusion as their second and beyond salvage treatment achieved CR (median, 274 days). CAR T-cell expansion was observed in 6 patients (n = 4 with fludarabine and cyclophosphamide conditioning, and n = 2 with cyclophosphamide conditioning), with a median peak expansion of 18 900 vector copy number (vcn) per mL (746-200 363 vcn/mL), compared with 48 451 vcn/mL (6819-2 320 302 vcn/mL) with the initial infusion (P = .15) (Figure 4). The pattern of CAR T-cell expansion after reinfusion varied. One patient (MSK36) had a higher median peak CAR T-cell expansion, whereas 3 patients (MSK17, MSK29, and MSK56) had comparable expansions compared with the initial infusion. After achieving CR, 1 patient underwent consolidative alloHSCT, but relapsed at 4 months; the other 2 patients subsequently relapsed within 3 months; and 1 patient died of CAR T-cell–associated complications.
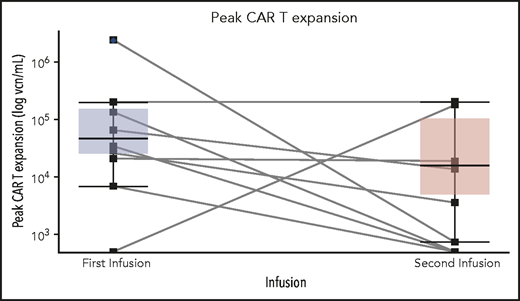
Peak CD19 CAR T-cell expansion comparison between the initial and second CAR T-cell infusion among 10 patients who had CAR T-cell retreatment. There was no significant difference between median peak CAR T-cell expansion between the first and second CAR T-cell infusion. Each solid line links the peak vcn/mL of CAR T cells of each individual patient at the first and second infusion. The top and the bottom of the boxplots reflect the interquartile ranges of CAR T-cell expansion. The thick line in each boxplot represents the median of peak CAR T-cell expansion.
Peak CD19 CAR T-cell expansion comparison between the initial and second CAR T-cell infusion among 10 patients who had CAR T-cell retreatment. There was no significant difference between median peak CAR T-cell expansion between the first and second CAR T-cell infusion. Each solid line links the peak vcn/mL of CAR T cells of each individual patient at the first and second infusion. The top and the bottom of the boxplots reflect the interquartile ranges of CAR T-cell expansion. The thick line in each boxplot represents the median of peak CAR T-cell expansion.
Discussion
Previous studies reported poor prognosis of patients with relapsed B-cell non-Hodgkin lymphoma and chronic lymphocytic leukemia after CD19 CAR T-cell therapy,9-11,16 but such comprehensive data for patients with B-ALL have not been published. To the best of our knowledge, our study is the first report that delineates this difficult-to-treat but increasingly more common patient population. Our study affirms poor outcomes of adult patients with persistent or progression of B-ALL after CD19 CAR T-cell therapy and highlights the need for innovative approaches to improve CAR T-cell efficacy and prevent relapse in adult ALL.
In our study, high disease burden before CAR T-cell therapy was the only significant predictor for progression post–CAR T cells, emphasizing the importance of pretreatment disease burden evaluation for proper risk stratification and prognostication. Notably, prior therapies including blinatumomab and inotuzumab did not affect relapse risk and survival after post–CAR T progression. The effect of prior exposure to CD19-targeted agents on subsequent CD19 CAR T cells is particularly concerning. Pillai et al17 reported inferior survival after CD19 CAR T-cell therapy in pediatric patients with B-ALL previously treated with blinatumomab. In contrast, data from the ZUMA-3 phase 1 trial in adult B-ALL and a study of large cell lymphoma showed that CD19 CAR T-cell efficacy was not compromised by previous treatments with blinatumomab and CD19-antibody drug conjugate, respectively.18,19 These conflicting data suggest further exploration is required to better define the impact of sequential CD19-targeted therapies in ALL, and CD19 expression should be assessed at the time of relapse in patients who received prior CD19-directed therapy. We also observed that severity of CRS and ICANS, including use of tocilizumab and steroids, did not affect the relapse risk, similar to findings from other studies.20-22 However, since the report of our initial study, CRS and ICANS management strategies have evolved over time with earlier use of tocilizumab and steroids, and our finding should be interpreted with caution as we await longer follow-up data from ongoing clinical studies of CAR T-cell therapies.
Owing to the long enrollment period of the study and no mandatory standard protocols for relapse post–CAR T cells, we observed wide ranges in management patterns of post–CAR T progression from chemotherapy to second infusion of CAR T cells. Overall, only 40% of the patients responded to the salvage treatments, and remission duration was short despite subsequent consolidative alloHSCT, with a median EFS of 5.8 months. There was no single regimen associated with superior survival. Notably, 7 (58%) of 12 patients achieved CR after blinatumomab or inotuzumab. Although limited by small numbers, our data suggest that antibody-based immunotherapies such as blinatumomab and inotuzumab can serve as an effective therapeutic strategy after CD19 CAR T failure. This finding is also supported by the study by Schuster et al,23 which reported 68% CR rates with mosunetuzumab, a CD3/CD20 bispecific monoclonal antibody, in patients with R/R B-cell lymphoma who did not respond to or relapsed after CD19 CAR T-cell therapy. In our study, reinfusion with CD19 CAR T cells resulted in a CR rate of 40%, similar to that reported with another CD19 CAR T-cell product in B-cell leukemia and lymphoma.24,25 However, long-term survival was limited, with only 2 of 10 patients who received CAR T-cell reinfusion remaining alive. CAR T-cell expansion after reinfusion was similar or higher compared with the initial infusion in 4 of 10 patients. There was a trend toward superior CAR T-cell expansion with fludarabine-containing lymphodepletion chemotherapy vs cyclophosphamide alone, but the small number of patients precluded a formal statistical analysis. We did not observe a clear association between choice of conditioning chemotherapy and response to the reinfusion.
The poor outcome of adult B-ALL progressing after CAR T-cell therapy likely reflects the heavily pretreated patient population in our cohort, with a median 3 prior lines of therapy and pre-CAR alloHSCT in 45% of the patients. These prior treatment histories and chemorefractory disease may partly explain the observed association between pre-CAR T bridging chemotherapy and worse OS with potential emergence of resistant leukemic clones from repeated chemotherapy exposure. Moreover, data from pediatric ALL showed that repeated exposure to cytotoxic agents can disrupt precursor T-cell repertoires, and in turn affect autologous CAR T attributes26,27 and compromise the efficacy of CAR T cells.28-30 As such, incorporation of CAR T-cell therapy in earlier lines of therapy and at lower disease burden may enhance the long-term efficacy in adult ALL, but better understanding of disease biology and its interaction with immune effector cells will be critical to further improve the promise of CAR T-cell therapy.
Our study has several unique strengths. It is the first and largest report presenting detailed data of adults with B-ALL who progressed after CD19 CAR T-cell therapy, including subsequent therapeutic management, outcomes, and long-term follow-up. Our study includes analysis of patients who received blinatumomab or inotuzumab after CD19 CAR T cells with encouraging responses, albeit limited by small numbers. Given the paucity of data of effective post-CAR T-salvage treatment options and anticipated approval of CD19 CAR T products in adult ALL, our findings provide more fundamental insights into an optimal sequence of treatments in B-ALL. However, the relatively small sample size and heterogeneous post-CAR T-salvage treatments in our cohort limit the statistical power to determine whether certain post–CAR T treatments are associated with better outcomes. This study also only reflects the experience with R/R disease after autologous 19-28z CAR T-cells, and characteristics of post–CAR T progression in recipients of other CAR T-cell products may be distinct. Lastly, our study focused on adult patients, and clinical experience in pediatric B-ALL patients may differ.
In summary, our study identifies adult patients with B-ALL at high risk of disease progression after CD19 CAR T-cell treatment and highlights the poor prognosis of those patients who progress after the therapy that may serve as a historical standard by which to evaluate post-CAR T therapeutic approaches. Future studies should prospectively investigate optimal sequence of antibody-based and cellular immunotherapies in B-ALL and develop strategies to reduce relapse and improve survival following CD19 CAR T-cell therapy, including the use of alternative immune effector cell source and novel CAR constructs with enhanced signaling domains or armored cytokines.31-33
Acknowledgments
The authors thank the patients who participated in this trial and their families, as well as the staff of the Cell Therapy and Cell Engineering Facility.
K.W. receives salary support from the Parker Institute for Cancer Immunotherapy at Memorial Sloan Kettering Cancer Center and King Chulalongkorn Memorial Hospital Thai Red Cross Society, Chulalongkorn University. This work received support from the National Institutes of Health, National Center for Advancing Translational Sciences (grant UL1TR00457; M.B.G.) and the National Cancer Institute (grant P30 CA008748).
Authorship
Contribution: K.W. and J.H.P. designed the study and wrote the manuscript; K.W. and J.H.P. collected the data and conducted the analysis; I.R., X.W., B.S., K.J.C., M.R., P.G.M., M.G.B., E.F.H., C.D., M.L.D., M.S., and R.J.B. participated in data collection; J.R.F. and M.G. conducted the statistical analysis; K.J.C., M.R., P.G.M., M.G.B., E.F.H., C.D., M.L.D., M.S., R.J.B., and J.H.P. took care of the patients; and all authors reviewed and approved the manuscript.
Conflict-of-interest disclosure: I.R. discloses stock or ownership interest, consulting or advisory role, research funding, and patents and royalties, all from Juno Therapeutics. K.J.C. has received research support from Juno Therapeutics and Novartis; and has consulted, served on advisory boards, and participated in educational seminars for Juno Therapeutics, Novartis, and Mesoblast. P.G.M. receives research funding from Sellas Life Sciences. M.B.G. received research support from Amgen and Actinium; and serves on the advisory board for Sanofi. M.L.D. reports receiving grants and licensing fees from Atara Biotherapeutics and CRISPR; research grants from Novartis and Kite/Gilead; and stock or stock options from Adaptive Biotechnologies, Precision Biosciences, and Bellicum outside the submitted work. M.S. receives research funding from Atara Biotherapeutics, Fate Therapeutics, and Takeda; and reports a patent/loyalty with Juno Therapeutics through MSKCC. R.J.B. provides advisory roles for Gracell Biotechnologies, AstraZeneca, and BMS/Juno Therapeutics; and reports royalties from BMS/Juno Therapeutics and Caribou Biosciences. J.H.P. receives funding from the Conquer Cancer Foundation of ASCO, a Leukemia and Lymphoma Society Career Development Grant, the Geoffrey Beene Cancer Foundation, a National Comprehensive Cancer Center Young Investigator Award, and an American Society of Hematology Scholar Junior Faculty Award; and has consulted and provided an advisory role for Amgen, Novartis, Kite Pharma, Incyte, Allogene, Autolus, Intellia, Artiva, AstraZeneca, Pfizer, Takeda, and Servier. The remaining authors declare no competing financial interests.
Correspondence: Jae H. Park, Leukemia Service, Division of Hematologic Malignancy, Memorial Sloan Kettering Cancer Center, New York, NY 10065; e-mail: parkj6@mskcc.org.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal