


Abstract
Nongenetic heterogeneity, or gene expression stochasticity, is an important source of variability in biological systems. With the advent and improvement of single molecule resolution technologies, it has been shown that transcription dynamics and resultant transcript number fluctuations generate significant cell-to-cell variability that has important biological effects and may contribute substantially to both tissue homeostasis and disease. In this respect, the pathophysiology of stem cell-derived malignancies such as acute myeloid leukemia and myelodysplastic syndromes, which has historically been studied at the ensemble level, may require reevaluation. To that end, it is our aim in this review to highlight the results of recent single-molecule, biophysical, and systems studies of gene expression dynamics, with the explicit purpose of demonstrating how the insights from these basic science studies may help inform and progress the field of leukemia biology and, ultimately, research into novel therapies.
Introduction
Cellular identity is encoded by the repertoire of genes expressed and repressed by a cell. These baseline patterns of gene expression, as well as the functional modules executed by the cell, are organized in networks. The performance of all transcription networks is contingent on the selective participation of the genome in a spatiotemporally coherent manner. In that regard, transcription factors (TF) are essential players in the maintenance of cellular identity through the specific and regulated licensing of loci in the genome. Although developing a quantitative understanding of TF dynamics and their impact on transcriptional networks is an area of obvious interest to developmental biology, aberrant networks are also a defining and ubiquitous feature of oncogenesis. In particular, dysregulation of normal differentiation networks and maintenance of a progenitor-like state seems to be prevalent in acute leukemogenesis.1-9 Indeed, as the high degree of genetic heterogeneity found both within and between patients with myelodysplastic syndromes and acute leukemias continues to be appreciated,10-13 deregulation of essential hematopoietic TF such as CEBPA, RUNX1, MEIS1, HOXA9, GATA2, and PU.1, may be the unifying pathological hallmark of these notoriously recalcitrant tumors rather than any particular genetic event.14-22 Elucidating the operational rules of normal hematopoietic differentiation networks will therefore affect our understanding of tumor pathogenesis and may inform novel therapeutic strategies.
Over the past few decades, technological developments have facilitated the study of gene expression at increasingly finer resolution. The forefront of these techniques has enabled the investigation of single cell behaviors, and at times even the study of single messenger RNA (mRNA) and protein molecules in living cells. When combined with powerful computational tools, machine learning algorithms, and theoretical insights from systems biology, these studies have provided an unprecedented and rapidly evolving understanding of life within the cell. This insight has, in many cases, demonstrated substantial intrinsic heterogeneity, or stochasticity, in the behavior of genetically identical cells in homogeneous conditions. Although these results are largely congruent with predictions made many decades ago by theoretical biologists and physicists,23-25 how to reconcile the complicated, even counterintuitive, picture emerging of life at the single-cell level with our notion of robust steady-state hematopoiesis is a challenge. Similarly, these studies present a major hurdle for the field in understanding the pathogenesis and the therapeutic targeting of malignancies such as myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML).
In this review, we will highlight the results of recent in situ, single-molecule, biophysical, and systems studies of transcription dynamics. The goal is to demonstrate how the results of these studies may require a reformatting of our understanding of leukemogenesis, particularly at the single-cell level. Indeed, as the oncogenicity imputed to TF “misexpression” is based on the premise that TF regulate their target genes in a concentration-dependent fashion, a quantitative model of TF expression and subsequent gene regulation is needed to ultimately understand AML pathogenesis.
Deregulation of transcription factors in AML
The complex cascade of reactions involved in “gene expression” offers multiple avenues by which TF levels and/or activity may be manipulated (Figure 1). Before delving into the major findings from single-cell gene expression studies, we will present a nonexhaustive survey of how hematopoietic transcription factors are known to be deregulated in AML.
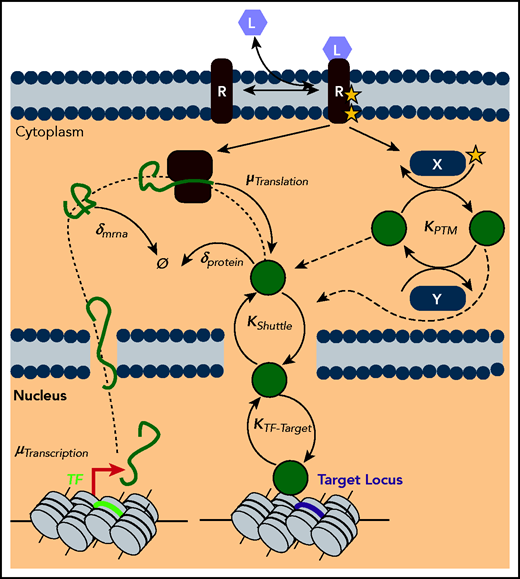
Gene expression as a multilevel reaction cascade. Schematic of the complex reaction cascade of gene expression. Deregulation of TF levels or activity can putatively occur along any step in the pathway, from synthesis steps such as transcription (µTranscription) and translation (µTranslation), to degradation steps such as the decay of mRNA (δmrna) and protein (δprotein), to dynamic processes such as posttranslational modifications by ligand activated pathways (KPTM), to shuttling dynamics (KShuttle), and finally to target locus search and binding (KTF-Target).
Gene expression as a multilevel reaction cascade. Schematic of the complex reaction cascade of gene expression. Deregulation of TF levels or activity can putatively occur along any step in the pathway, from synthesis steps such as transcription (µTranscription) and translation (µTranslation), to degradation steps such as the decay of mRNA (δmrna) and protein (δprotein), to dynamic processes such as posttranslational modifications by ligand activated pathways (KPTM), to shuttling dynamics (KShuttle), and finally to target locus search and binding (KTF-Target).
Transcriptional deregulation
The classic example of transcriptional deregulation of TF comes from one of the earliest identified AML oncogenes, the onco-fusion protein PML-RARα. In a series of seminal studies, it was determined that this fusion gene, the putative driver of acute promyelocytic leukemia, was actually the fusion of 2 TFs. As such, its pathogenicity is presumptively derived from its ability to deregulate the transcription network of the promyelocyte genome.3,26,27 More recently, the oncogenicity of another well-studied class of onco-fusion proteins, the MLL rearrangements, was demonstrated to be due to the mislocalization of the activating histone methyltransferase DOT1L, leading to increased RNA polymerase processivity genome wide and the overexpression of genes typically silenced during terminal myelopoiesis.28-34 In particular, MLL-AF9–mediated leukemogenesis in mouse models is thought to be dependent on the ability of this fusion to drive the aberrant expression of the HOX-related TF genes, Meis1 and Hoxa9, leading to a more primitive, blast-like state known as a leukemic granulocyte macrophage progenitor.35 It was also demonstrated that retroviral overexpression of these TF alone is sufficient to transform normal human hematopoietic stem and progenitor cells (HSPCs).36 Within human patient samples, one of the most striking examples of TF transcriptional deregulation is the aberrant expression of the EVI1 oncogene in inv(3) AML. This inversion event, which has 1 of the worst prognoses of all recurrent AML genetic events,37 reorients the highly active GATA2 enhancer toward the stem cell TF EVI1, leading to significant misexpression of the latter TF.6,9 EVI1 overexpression subsequently leads to installation and enforcement of a stem cell transcriptional network by physically antagonizing the transactivation of lineage specifying TF such as PU.1, GATA1, and RUNX1. Furthermore, the subsequent downregulation of GATA2 potentiates the EVI1 phenotype in human AML cells,9 a finding consistent with studies showing that haploinsufficiency of Gata2 accelerates Evi-1 driven AML pathogenesis in mice.38
Direct transcriptional repression of TF is also a hallmark of many AML etiologies. For instance, the onco-fusion RUNX1-ETO, generated by the t(8;21) translocation, is thought to induce leukemia in part by the repression of CEBPA transcription and PU.1-dependent transcriptional activation,39 leading to a block in terminal granulocyte differentiation.40 RUNX1-ETO has also been shown to directly repress the expression of the tumor suppressor TF, p14(ARF).41 Another powerful example of how direct transcription deregulation of TF can precipitate AML includes the myriad of ways in which the master regulatory myeloid TF PU.1 is affected during leukemogenesis. This deregulation occurs at both the level of changing PU.1 concentration, as well as through direct uncoupling of the PU.1 gene regulatory function at transcriptional targets (Figure 2A). One example of the former is the murine model of spontaneous AML generated by loss of the −14 kb upstream regulatory element of the Spi1 (PU.1) gene.8,42 Loss of this upstream regulatory element leads to a roughly 80% reduction in the mRNA and protein level of PU.1 in hematopoietic stem cell and myeloid progenitors, ultimately precipitating a fulminant AML with virtually 100% penetrance. Moreover, heterozygous loss of this enhancer on a mutagenic, mismatch repair-deficient background also generates MDS and progression to AML in ∼70% of mice.43 This finding is paralleled in human MDS and AML pathogenesis, with roughly 50% to 70% of tumors downregulating this key TF via a variety of mechanisms, including mutation of upstream regulators such as GFI1b and RUNX1, NPM1c mutations that inhibit PU.1 shuttling into the nucleus, or through sequestration and physical blocking of PU.1 protein by oncoproteins such as RUNX1-ETO.44-51 Additionally, it appears that mutations in the cohesin complex, particularly Stag2, limits PU.1 ability to access and activate genes required for terminal differentiation.52 Finally, PU.1 heterozygous mutations are found in a subset of MLL rearranged leukemias, with subsequent downregulation of PU.1 target genes46 (Figure 2B).

Multiple pathways of TF deregulation in AML and MDS. (A) TFs modify the output of target genes by influencing the reaction propensities of various steps in transcription in a manner proportional to some function of TF concentration. These functions can take on a variety of shapes, depending on the number of binding sites and the higher order complexes that a given TF is involved in at a target locus. Cooperativity at a locus produces gene regulatory functions (GRF) that are sigmoidal in shape. Although not mutually exclusive phenomena, in a simplistic sense deregulation can occur either through reducing the amount of TF produced or by changing the GRF shape through changing the degree of cooperativity. In the case of gene activation, this causes different effects at the level of single cells. In the former case (left), lower TF concentrations lead to less transcription of a target at the single-cell level. In the latter case (right), the amount of TF needed to sufficiently activate a target is increased, thereby reducing the number of cells which achieve the “threshold” concentration of TF. (B) Specific etiologies whereby the master myeloid TF PU.1 is deregulated in AML and MDS. Citations found within the main text. DBD, DNA-binding domain; PEST, Pro-Glu-Ser-Thr rich domain; TAD, transactivation domain.
Multiple pathways of TF deregulation in AML and MDS. (A) TFs modify the output of target genes by influencing the reaction propensities of various steps in transcription in a manner proportional to some function of TF concentration. These functions can take on a variety of shapes, depending on the number of binding sites and the higher order complexes that a given TF is involved in at a target locus. Cooperativity at a locus produces gene regulatory functions (GRF) that are sigmoidal in shape. Although not mutually exclusive phenomena, in a simplistic sense deregulation can occur either through reducing the amount of TF produced or by changing the GRF shape through changing the degree of cooperativity. In the case of gene activation, this causes different effects at the level of single cells. In the former case (left), lower TF concentrations lead to less transcription of a target at the single-cell level. In the latter case (right), the amount of TF needed to sufficiently activate a target is increased, thereby reducing the number of cells which achieve the “threshold” concentration of TF. (B) Specific etiologies whereby the master myeloid TF PU.1 is deregulated in AML and MDS. Citations found within the main text. DBD, DNA-binding domain; PEST, Pro-Glu-Ser-Thr rich domain; TAD, transactivation domain.
Posttranscriptional deregulation
Although typically studied at the level of transcription, posttranscriptional deregulation is also known to occur in AML. This has been best described for the master granulocyte TF, CEBPA. CEBPA is translated as 2 major polypeptides, p30 and p42, where p30 can inhibit p42-mediated gene activation by competing for binding sites in the genome.53 Importantly, the selection of isoform expression is sensitive to eIF2 and eIF4e levels and consequently, mTOR signaling. Intriguingly, although frameshift mutations in CEBPA leading to exclusive p30 isoform expression have been described in ∼9% to 10% of AML cases, mTOR pathway perturbation has been demonstrated to be a hallmark feature seen in essentially all patients with AML.54-56
Beyond the level of a single TF, changes in global rate parameters of posttranscriptional reactions are also known to occur during leukemogenesis. One active area of research is focused on understanding how the variety of mutations in splicing factors57,58 frequently seen in both AML and MDS lead to pathology. These mutations, including in SRSF1 and U2AF1, are known to change pre-mRNA processing and alternative exon selection across many genes in the genome.57,58 Similarly, mutations in ribosomal genes can lead to a spectrum of ribosomopathies, with some such as dyskeratosis congenita, Shwachman-Diamond syndrome, and 5q syndrome (owing to consequent haploinsufficiency of the RPS14 gene) having an increased propensity for leukemia.59 Furthermore, mutations in ribosomal subunits have been described across various cancer subtypes.60 Finally, deregulation of mRNAs posttranscriptionally by either base editing or through the altered expression of RNA binding proteins has been recently recognized as having a critical impact on gene expression61-67 (this emerging field in leukemia research is addressed in a companion review). Although systems-level analyses will be required to fully understand how these aberrations promote leukemogenesis, given the typically short mRNA and protein half-lives of TF genes,68 it is anticipated that posttranscriptional deregulation will have an enormous impact on the concentration and stoichiometry of these factors.
Finally, 1 important mechanism for TF deregulation in leukemogenesis is through changes in the cellular distribution of TF molecules. For instance, NPM1 mutations, which occur in ∼35% of primary AML patients,69 are thought to confer pathogenicity through the cytoplasmic sequestration of myeloid regulatory factors, including PU.1.70 Our group recently described a role for the overexpression of MDMX in human AML, which subsequently leads to p53 sequestration.71 Additionally, the signaling pathways frequently found to be deregulated in leukemia communicate their aberrant signals through nuclear translocation of responsive TF such as ERK and STAT proteins (see previous work72 for a comprehensive review of this topic). Finally, 1 well-known mechanism of leukemogenesis, particularly in core-binding factor mutant AML, is the direct antagonism of lineage specifying TF,39,40,73 thereby leading to an uncoupling of TF levels and target gene responses.
Quite clearly, a number of distinct mechanisms have been described for how leukemia-initiating events generate and maintain a leukemic state. Although the exact details may vary, a hallmark seen in essentially every cytogenetic or mutational subgroup is the deregulation of TF behavior and/or expression. In that sense, TF deregulation may represent a unifying principle of leukemogenesis. As highly powerful analytical techniques and technologies with molecular resolution begin to be used in the study of leukemia, our understanding of how this process plays out at the molecular level in single cells is going to be a reality in the coming years. In the next section, we provide an overview of some of the basic findings derived from these types of technologies and what their impact may be on the future of leukemia research.
Gene expression at the molecular level
Although the studies listed have been foundational to our understanding of leukemogenesis, ensemble assays of gene expression inherently blur nongenetic variability between cells. This can be problematic when studying the pathogenesis of clonal diseases such as AML and MDS that arise from subpopulations of HSPCs,10,11 particularly given the lack of fully specific and sensitive markers to define leukemic stem cells. For one, the signal from the pathogenic process in question is diluted by signal from normal HSPCs. Arguably more important, however, is that even for identical cells in identical conditions, heterogeneity is a natural and unavoidable consequence of gene expression that has been documented at every level of the evolutionary tree.74-81 This heterogeneity, or stochasticity, derives from the physical nature of gene expression: the reactions constituting “gene expression” are a series of punctuated steps involving low copy numbers of reactants, in poorly mixed, partitioned reaction chambers within the cell. Moreover, capturing this variability is not simply an academic exercise. Indeed, this stochasticity provides critical information about the dynamics and potential regulatory systems of the process under study.82,83 As such, quantitatively studying gene-expression stochasticity, particularly in context of TF dynamics, is an essential “next step” in the field’s investigation of leukemia pathogenesis and treatment. To that end, in the following sections we focus on how molecular randomness and biophysical forces influence the behavior of TF in the cell.
Transcriptional bursting
As the initial step in gene expression, the reaction kinetics of transcription have been a focus of intense investigation. The advent of in situ single-molecule imaging tools has provided the necessary resolution for the quantitative analysis of those kinetics. The first of these tools was single-molecule mRNA fluorescence in situ hybridization (smRNA-FISH), developed by Femino and colleagues in Rob Singer’s group in 1998.84 This transformative technology allows for the detection of individual molecules of mRNA while maintaining spatial information within the cell. This key property of smRNA-FISH enables one to not only accurately capture the probability distribution of a gene across a cell population, but also to facilitate direct observation of nascent transcription. When combined with computational modeling approaches, this has allowed for the inference of the underlying transcriptional rate parameters of a gene. The first of these efforts was performed in a seminal study by Arjun Raj and colleagues, who studied the transcriptional kinetics of a tetracycline inducible minigene using smRNA-FISH and a theoretical framework known as the random telegraph model.85,86 This work provided the first inference of key rate constants of an endogenous gene, including the rate of gene activation, gene inactivation, and polymerase initiation rates. Similar approaches have been subsequently used to study the transcription of genes from a diverse repertoire of species and situations, including bacteria,87 yeast,78 Drosophila,88 and mammalian cells,89-92 including recently in primary HSPCs from mice.93 Irrespective of the system studied, the common feature found is that transcription is a discontinuous (ie, bursting, phenomenon), whereby individual loci undergo cycling between intervals of productive transcription followed by longer periods of inactivity (Figure 3A). For instance, we have recently found that although the majority of primary HSPCs express the key TF PU.1, Gata1, and Gata2 genes at the mRNA level, active transcription is surprisingly rare, with only 10% to 20% of cells expressing at any given moment.93 Even in unipotent populations of cells typified by high levels of either Gata TF or PU.1 (ie, megakaryocyte-erythroid progenitor and granulocyte macrophage progenitor cells), these frequencies only increased to ∼40% and 20%, respectively. For low-copy-number bursts or mRNAs with shorter half-lives, features typical of many TF mRNAs,93,94 these temporal fluctuations in transcriptional activity can lead to markedly dispersed single-cell mRNA probability distributions.91,95 Furthermore, these observations with smRNA-FISH have been validated with live cell imaging studies of nascent transcription using the MS2 mRNA tagging system.96 In this technique, an array of stem loop sequences derived from the MS2 bacteriophage RNA genome are incorporated into the 3′ untranslated region of a gene of interest. By coexpressing a fluorescently tagged MS2 viral coat protein, which binds these stem loops, in the cell, single puncta corresponding to single mRNA are readily visualized. Using this approach, live cell dynamics of transcriptional bursting can be directly measured.97 Consistent with the data derived from smRNA-FISH, MS2-tagged endogenous alleles undergo cyclic bursting interspersed between long “OFF” periods in gene activity.71,97-105 For instance, a recent landmark paper from Dan Larson’s group demonstrated that bursts in the expression of the estrogen response gene TFF1 can be separated by inactivate periods lasting on the order of days, even after induction with estradiol.103
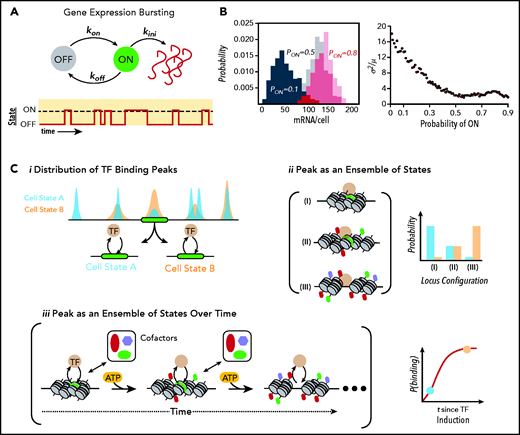
Gene expression at the molecular level. (A) Schematic of a simple stochastic switch whereby a molecule of interest (ie, a gene, mRNA, or protein) undergoes cyclical activation and deactivation reactions, thereby rendering the molecule capable or refractory to an additional reaction. In the example of promoter cycling and transcription, this leads to probabilistically distributed bursts of transcription followed by periods of quiescence. (B) Simulated data showing how for a simple 1-species system produced in bursts and undergoing first-order decay, the probability of firing is directly related to the heterogeneity within a population of identical cells. Histograms on the left show the probability distributions for a gene with a probability of being “ON” (Pon) of 0.1 (black), 0.5 (gray), and 0.8 (red). Plot on the right demonstrates how population noise (captured with the Fano Factor, or the variance/mean) changes as a function of (Pon). A Poissonian distribution (Fano = 1) is the minimal noise observed within biological systems.83 For many TFs, Pon is ∼0.1 to 0.3.93 (C) Relationship of ensemble assays of TF-binding activity to actual behavior of molecules. (i) From the binding distributions derived from ensemble studies such as chromatin immunoprecipitation sequencing, one can conclude that at the green locus, cells in state B immunoprecipitated the locus more frequently than in state A, where states could be defined by a variety of measures including cell surface proteins, cell-cycle position, or metabolic profiling. However, the ensemble studies cannot determine whether this is due to changes in the configurations of chromatin states (ii), each with different affinities for the TF, or from differences in the temporal evolution of the locus (iii), or some combination. Of note, as indicated in the bottom right of iii, state A and state B may lie on a temporal continuum rather than representing discrete entities. This evolution in time is based on energy-dependent modification of the locus by epigenetic enzymes licensed to the site by the TF. Importantly, this implies that cells with identical concentrations of TF could have very different binding patterns (and therefore target gene activity) solely because of each cell’s position on the time curve. This ambiguity obviously complicates the interpretation of ensemble studies of TF binding and how such binding influences target locus expression.
Gene expression at the molecular level. (A) Schematic of a simple stochastic switch whereby a molecule of interest (ie, a gene, mRNA, or protein) undergoes cyclical activation and deactivation reactions, thereby rendering the molecule capable or refractory to an additional reaction. In the example of promoter cycling and transcription, this leads to probabilistically distributed bursts of transcription followed by periods of quiescence. (B) Simulated data showing how for a simple 1-species system produced in bursts and undergoing first-order decay, the probability of firing is directly related to the heterogeneity within a population of identical cells. Histograms on the left show the probability distributions for a gene with a probability of being “ON” (Pon) of 0.1 (black), 0.5 (gray), and 0.8 (red). Plot on the right demonstrates how population noise (captured with the Fano Factor, or the variance/mean) changes as a function of (Pon). A Poissonian distribution (Fano = 1) is the minimal noise observed within biological systems.83 For many TFs, Pon is ∼0.1 to 0.3.93 (C) Relationship of ensemble assays of TF-binding activity to actual behavior of molecules. (i) From the binding distributions derived from ensemble studies such as chromatin immunoprecipitation sequencing, one can conclude that at the green locus, cells in state B immunoprecipitated the locus more frequently than in state A, where states could be defined by a variety of measures including cell surface proteins, cell-cycle position, or metabolic profiling. However, the ensemble studies cannot determine whether this is due to changes in the configurations of chromatin states (ii), each with different affinities for the TF, or from differences in the temporal evolution of the locus (iii), or some combination. Of note, as indicated in the bottom right of iii, state A and state B may lie on a temporal continuum rather than representing discrete entities. This evolution in time is based on energy-dependent modification of the locus by epigenetic enzymes licensed to the site by the TF. Importantly, this implies that cells with identical concentrations of TF could have very different binding patterns (and therefore target gene activity) solely because of each cell’s position on the time curve. This ambiguity obviously complicates the interpretation of ensemble studies of TF binding and how such binding influences target locus expression.
Finally, these bursting behaviors are not restricted to mRNAs. Indeed, live cell imaging of translation has indicated that these reactions also occur in discrete bursts. In a series of seminal, concurrent papers, 4 groups demonstrated that translation of an mRNA is also an infrequent event, with the majority of mRNAs not translating at any given moment.106-109 For instance, it was demonstrated that for the Actb gene in murine neurons, only 10% to 30% of mRNAs were translating at any given moment in the cell. Furthermore, once an mRNA was engaged by the polysome, roughly 4.5 nascent peptides were produced before translation termination.110 Although these techniques are still relatively new and require further study, when taken together with results at the transcriptional level, there is a clear demonstration that bursting phenomena are intrinsic properties of multiple levels of the central dogma, effectively guaranteeing nongenetic, population heterogeneity in the level of any biochemical species of interest in the cell (Figure 3B).
Subcellular TF dynamics
Although it is evident that the intracellular concentration of a TF influences transcriptional networks, live cell protein imaging has demonstrated significant complexities in the processes of TF localization, target search, and locus binding. These dynamic processes impact target gene expression and transcriptional networks in ways that are only just now starting to be appreciated. Nevertheless, there is growing evidence that the spatiotemporal behavior of TF proteins is an important component of gene regulation and an additional layer of nongenetic heterogeneity in single cells.
TF shuttling between the nucleus and cytoplasm is currently the best studied dynamic behavior. In both yeast and mammalian cells, it appears that the rate and amplitude of these subcellular translocations impart meaningful information to the transcriptome not accounted for by concentration alone.111,112 For instance, differences in shuttling rates of stress TF in yeast can lead to differential output of coregulated genes,113 and differential p53 responses to γ-irradiation and ultraviolet genotoxic stress are encoded by differences in shuttling behaviors rather than changes p53 concentration.114-116 Similar shuttling behaviors have also been described for NF-κB,117 ERK2,118,119 and SMAD4.120 These studies and others demonstrate mechanistically that subcellular protein dynamics may provide critical information to the transcriptome that is independent of the TF concentration at a given moment. Of note, similar effects have recently been reported to be a key step in the oncogenesis of NPM1c mutations, with changes in the shuttling capacity of the master transcription factor PU.1.70 As such, this relatively unexplored area of gene deregulation may have important roles in AML pathogenesis.
Another area of TF biology markedly advanced by single-molecule resolution imaging studies is our understanding of how TF find and bind to target genes. Indeed, the facilitated diffusion models derived from bacterial studies,121 where TF passively explore 3-dimensional space followed by constrained 1-dimensional sliding on DNA, physically cannot work in eukaryotes given the enormity of the genome as well as the chromatin-derived energy barriers to sliding.122 In recent years, the application of single protein imaging in live cells has facilitated direct observation of TF search paths through the mammalian nucleus. The results have been striking and at times counterintuitive. For instance, although the general transcriptional amplifier MYC123 and the transcriptional elongation complex P-TEFb124 are postulated to work in concert, they explore the nuclear space with decidedly different modes: MYC globally surveys the entire volume of the nucleus whereas P-TEFb undergoes constrained diffusion.125 Similarly, CTCF and Rad21, 2 integral and cooperating proteins controlling the topology of the eukaryotic genome, also have markedly different search dynamics: although a diffusing CTCF typically searches for and binds to a cognate site within 1 minute, Rad21 requires on average 33 minutes of diffusion before rebinding.126
One somewhat surprising finding from the studies listed here is not just the differences in search strategies used by different regulatory proteins, but also the instability of binding once a target has been identified. Indeed, CTCF was found to bind to its cognate binding site for only 1 minute on average, whereas Rad21 bound for 22 minutes.126 These data indicate that the stable looping structures assumed through -omics approaches are actually maintained through a flux of protein. Even more remarkably, it appears that for many nuclear proteins, including SOX2,127,128 the Mediator complex, RNA polymerase II129 and BRD4,130 binding times are on the order of seconds, rather than minutes, indicating that most regulatory complexes are transient hubs maintained through dynamic binding and unbinding of factors to the genome. Although preliminary, our group has also recently discovered similar properties in the hematopoietic TF PU.1 and Gata1, with each having stable binding occurring in only 5% of all nuclear TF molecules at a given time and with residence times of only several seconds during each binding event.
Quantitative gene regulation
Another consideration that is exceedingly nontrivial in light of the preceding section is determining how a TF actually regulates a target locus. In principle, mammalian TF are known to either activate or repress target genes through the recruitment of enzymes to chromatin. This recruitment then changes the local energy landscape through covalent modification of histones and/or DNA, ultimately facilitating or hindering subsequent transcription131,132 (see the companion review for more on this topic). Although this model is typically assumed, implicit in this description is the consumption of energy, which means that the system out of thermodynamic equilibrium. This has at least 2 important implications for the leukemia field.
First, whereas the interaction of a TF and a target locus are typically viewed from the perspective of a static DNA substrate being bound by protein (Figure 3Ci), the microscopic reality of this interaction is actually much more nuanced. Indeed, the inclusion of epigenetic reactions actually means that the DNA-chromatin template could either be in a number of energetically different configurations within the same “cell state,” with consequently different binding activities by the TF that are stable properties for the locus over the cell’s lifespan, or it may reflect changes in the locus over time within the same cell (Figure 3Cii-iii).133,134 Although these phenomena are clearly not mutually exclusively, in the restricted case of simple temporal evolution, the binding affinities between a TF and a target locus will necessarily change over time. Given the probabilistic nature of binding and unbinding, as well as the short time scales over which these interactions take place, it is therefore evident that identical cells with similar TF concentrations could demonstrate markedly different behaviors over time through chance alone. Although this may seem to be a purely academic distinction, this directly affects the validity of the derived gene regulatory function87,133 linking the concentration of a TF to target gene expression. From a practical perspective, it indicates that contemporaneous transcriptional activity of a gene may be better correlated with the temporal integration of TF-binding events, rather than the current TF-binding peaks. This is likely to be particularly relevant for TF that are expressed at low, but fluctuating, levels beginning in early stem cells such as PU.1, Runx1, and Gata2. A second, related issue comes from the observation that many TFs appear to undergo stochastic state transitions between low and high expression states over the course of many cell divisions during differentiation.90,91,93,135,136 The number of these transitions, the time spent in each state, and the half-lives of the covalent modifications made at the TF’s target loci all have direct effect on the transcriptional network of the cell.
Taken together, these 2 effects of nonequilibrium dynamics necessitate the study of transcriptional network evolution over time, rather than through single snap shot analyses.134 At a technical level, this consideration ultimately requires that the root, leukemia-initiating cell populations are defined and studied. Recent advances in single-cell technologies, single-molecule imaging, and clonal tracing will be critical tools in this regard.90,93,137-141 Furthermore, advances in information theory,142,143 noise-control theory,144-147 and multitalk evolution theory148,149 will be of particular importance in making analytical sense of these complex data sets. Ultimately, these tools, both technical and analytical, should help formulate a more comprehensive, unified understanding of MDS and AML (and other cancers) and point the way toward novel therapeutic options.
Defining the leukemic state and therapeutic resistance
The aforementioned studies significantly complicate our understanding of transcriptional networks, how they emerge, and how TF regulate them. For one, transcriptional stochasticity leads to probability distributions that are significantly dispersed and typically dominated by low molecular copy numbers on the order of tens of mRNAs and hundreds to a few thousand protein molecules per cell. Second, even after gene expression reactions are completed, the behavior of these TF protein molecules is complex, relying heavily on protein–protein interactions and dynamic posttranslational modifications, in addition to the canonical interactions they make with the genome. Finally, all of these fluctuating sources of regulatory information ultimately must be “read out” and culminate in a stereotypical pattern of gene expression known as the transcription network. Undoubtedly, determining how these processes reliably transmit information to support robust tissue homeostasis is one of the major outstanding questions of molecular and developmental biology. By extension, how this putatively noisy flow of information can be further corrupted to generate a leukemic state or leukemic network also remains to be fully defined.
One very possible scenario in light of this realization is that a defined leukemic state may not be an appropriate view of disease pathogenesis in AML and MDS. This viewpoint is built from the large body of literature that has carefully collected and cataloged the litany of disease-associated mutational variants or deregulated genes found within these tumors. From those mutations, the assumption has been that a network must be operating to maintain fitness of leukemic clones. However, this model has not be able to explain why so many mutations both within10,11 and across leukemia patients37 lead to largely phenotypically similar conditions. Indeed, a “unified” theory of leukemogenesis is still needed.
One alternative hypothesis is that leukemic cells or the leukemic network may be better understood as a set of “trapped” cellular states on the differentiation landscape normally supporting multilineage, hematopoietic differentiation, akin to a molecular purgatory from which the clone cannot escape (Figure 4A). In such a model, the leukemic state would effectively be a default state cells “fall into” when ≥1 critical network components are deregulated outside of their evolutionarily set operating window. Leukemogenicity would, in this situation, not be defined by a unique oncogenic program, but rather as the set of programs conducive to continued cell survival despite failure to progress along a differentiation trajectory. Importantly, the only requirement of such a default state is that the state is viable; it could comprise an innumerable number of substates driven through stochasticity of gene expression. Quite obviously, there will be a pressing need to concurrently define the boundaries within which “normal” hematopoietic network dynamics can operate to understand where in transcriptional space leukemic networks reside.

The leukemic state and disease progression. (A) Topology of normal hematopoietic differentiation with the “leukemic state” as a trapped basin in gene expression space. (B) Two models of therapeutic resistance in AML. In the genetic scenario, acquisition of resistance mutations (denoted with R+) in a polyclonal malignancy creates a selective pressure that leads to AML relapse. In the transcriptional state plasticity scenario, cells can occupy a diversity of cellular microstates within the “leukemic state” that make them more or less susceptible to clearance by chemotherapy. If these states are based on the expression of particular TF, then selected states could be inherited in relapse tumors. Importantly, these mechanisms are likely not mutually exclusive and are intrinsically dependent on one another.
The leukemic state and disease progression. (A) Topology of normal hematopoietic differentiation with the “leukemic state” as a trapped basin in gene expression space. (B) Two models of therapeutic resistance in AML. In the genetic scenario, acquisition of resistance mutations (denoted with R+) in a polyclonal malignancy creates a selective pressure that leads to AML relapse. In the transcriptional state plasticity scenario, cells can occupy a diversity of cellular microstates within the “leukemic state” that make them more or less susceptible to clearance by chemotherapy. If these states are based on the expression of particular TF, then selected states could be inherited in relapse tumors. Importantly, these mechanisms are likely not mutually exclusive and are intrinsically dependent on one another.
Beyond disease initiation, this view of leukemia gene expression states may also have an important role in therapy resistance. Indeed, network plasticity derived from nongenetic heterogeneity has been demonstrated in other tumors to generate population fitness. In both neuroblastoma150 and melanoma,138,151 stochastic transitions in the tumor’s gene expression network facilitate the escape of a subpopulation of tumor cells when challenged with chemotherapy. Foundational studies in metastases also have shown that heterogeneity within the primary tumor is central to the metastatic capacity of the tumor.152 Determining whether such mechanisms, as opposed to purely genetic mechanisms, are operating during leukemia relapse will be a critical step in the coming years (Figure 4B).
Summary/conclusion
In conclusion, recent studies at the level of single molecules and in single cells have shed new insight into the complexities of gene expression. In light of those studies, we believe that it will be particularly important to consider the molecular, spatial, and temporal resolution needed to study gene expression changes during leukemogenesis, particularly when the time scales (eg, binding times of TF, bursting frequencies) and the molecular thresholds (eg, the number of TF molecules needed to influence a network) operating in these systems remain poorly defined. We believe the technologies listed here will be critical in this regard, particularly when the results of those studies are used in parallel to the exciting advances coming from whole genome level, including single-cell RNA-sequencing,137,140,153-156 assay for transposase-accessible chromation sequencing,157,158 chromatin capture techniques,52 and chromatin immunoprecipitation seqencing.73,159 Moreover, they can be used to test the predictions made via elegant network analyses of hematopoiesis and leukemia.160-163 For one, tools such as single-molecule FISH will be vitally important toward understanding both the magnitude and source of gene expression stochasticity in primary hematopoietic cells. Live cell studies of transcription could also be a useful tool for investigating how different epigenetic aberrations seen in AML lead to changes in transcriptional kinetics. Single-protein imaging experiments in live cells could provide critical measurements of actual search times and binding times of nuclear proteins such as TF, transcriptional machinery, and cohesin complex components, which will help shape our understanding of chromatin dynamics, how combinatorial control of loci plays out in live cells, and how these dynamics are altered in leukemia. As with all technical approaches, it will be important to consider the biological and physical plausibility of models derived from these studies a priori,133,146,147,164,165 particularly in light of the wide range of single-cell behaviors that are evidently tolerated during normal hematopoiesis. Finally, it is also clearly evident that any of these tools (either alone or in combination) should also play a pivotal role in the next generation of high-throughput screening technologies in the search for novel therapeutics or therapeutic strategies. Indeed, the combination of high-resolution, single-cell technologies with systems analyses has recently yielded fundamental insights into how pharmacology can affect dynamic signaling processes in the cell.119 It behooves the leukemia community to begin applying similar experimental approaches in the quest for novel antileukemic agents. A particularly alluring prospect would be to use these tools to revisit previously developed compounds that, although not necessarily efficacious as single agents, limit the gene expression state space leukemia cells can occupy and possibly increase the potency of current standard of care regimes.
In summary, the advent of single-molecule gene expression studies has been a watershed moment for our understanding of life inside the cell. Those studies have demonstrated that stochasticity imparts an enormous influence on the process of gene expression. It therefore follows that defining “gene deregulation” may be much more a complicated process than initially anticipated. Nevertheless, by integrating these tools in parallel to the critical whole-genome approaches being used in the field, it is the opinion of these authors that there will be significant progress in our understanding of leukemia pathogenesis in the coming years. This progress should hopefully open new therapeutic avenues into the treatment of these recalcitrant malignancies.
Acknowledgments
J.C.W. is supported by grants from the National Institutes of Health, National Institute of General Medical Sciences (F30GM122308 and T32GM007288). U.S. is supported by grants from the National Institutes of Health, National Cancer Institute (R01CA217092 and P30CA013330).
Authorship
Contribution: J.C.W. and U.S. wrote the paper.
Conflict-of-interest disclosure: U.S. has received research funding from GlaxoSmithKline, Bayer Healthcare, Aileron Therapeutics, and Novartis; has received compensation for consultancy services and for serving on scientific advisory boards from GlaxoSmithKline, Bayer Healthcare, Celgene, Aileron Therapeutics, Stelexis Therapeutics, and Pieris Pharmaceuticals; and has equity ownership in and is serving on the board of directors of Stelexis Therapeutics. J.C.W. declares no competing financial interests.
Correspondence: Ulrich Steidl, Albert Einstein College of Medicine, Chanin Building, Rm. 601-605, 1300 Morris Park Ave, Bronx, NY 10461; e-mail: ulrich.steidl@einsteinmed.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal