

Key Points
caADAMTS13 effectively restores blood flow following distal MCAo and reduces tissue hypoperfusion following ischaemia/reperfusion injury.
caADAMTS13 dissolves microvascular platelet aggregates and reduces neutrophil infiltration into the ischaemic tissue following stroke.

Abstract
Advances in our understanding of ADAMTS13 structure, and the conformation changes required for full activity, have rejuvenated the possibility of its use as a thrombolytic therapy. We have tested a novel Ala1144Val ADAMTS13 variant (constitutively active [ca] ADAMTS13) that exhibits constitutive activity, characterized using in vitro assays of ADAMTS13 activity, and greatly enhanced thrombolytic activity in 2 murine models of ischemic stroke, the distal FeCl3 middle cerebral artery occlusion (MCAo) model and transient middle cerebral artery occlusion (tMCAO) with systemic inflammation and ischemia/reperfusion injury. The primary measure of efficacy in both models was restoration of regional cerebral blood flow (rCBF) to the MCA territory, which was determined using laser speckle contrast imaging. The caADAMTS13 variant exhibited a constitutively active conformation and a fivefold enhanced activity against fluorescence resonance energy transfer substrate von Willebrand factor 73 (FRETS-VWF73) compared with wild-type (wt) ADAMTS13. Moreover, caADAMTS13 inhibited VWF-mediated platelet capture at subphysiological concentrations and enhanced t-PA/plasmin lysis of fibrin(ogen), neither of which were observed with wtADAMTS13. Significant restoration of rCBF and reduced lesion volume was observed in animals treated with caADAMTS13. When administered 1 hour after FeCl3 MCAo, the caADAMTS13 variant significantly reduced residual VWF and fibrin deposits in the MCA, platelet aggregate formation, and neutrophil recruitment. When administered 4 hours after reperfusion in the tMCAo model, the caADAMTS13 variant induced a significant dissolution of platelet aggregates and a reduction in the resulting tissue hypoperfusion. The caADAMTS13 variant represents a potentially viable therapeutic option for the treatment of acute ischemic stroke, among other thrombotic indications, due to its enhanced in vitro and in vivo activities that result from its constitutively active conformation.
Introduction
Recombinant tissue plasminogen activator (rtPA) stands alone as an approved thrombolytic therapy for the treatment of acute ischemic stroke (AIS), and the lack of safe, effective pharmacological alternatives is a prominent example of an unmet clinical need. A narrow therapeutic time window,1 significant associated risk of intracerebral hemorrhage,2 and efficacy that is dependent on thrombus composition3,4 have limited the successful application of rtPA to AIS patients. Several preclinical studies have identified a potential alternative thrombolytic therapy in the form of recombinant human (rh) ADAMTS13.
ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) is a plasma metalloprotease that targets the vascular adhesive glycoprotein von Willebrand Factor (VWF), regulating its multimeric size and its numerous prothrombotic and proinflammatory functions.5,6 The primary function of VWF is tethering of circulating platelets at sites of vascular damage; however, it has also been implicated in the recruitment of neutrophils7 and monocytes8 to the vascular endothelium, in the formation of tissue factor-expressing platelet-monocyte aggregates,9 and in the process of platelet “priming” of neutrophils.10 Early experimental studies using VWF−/− and ADAMTS13−/− mice confirmed the importance of the VWF/ADAMTS13 axis in the context of acute myocardial infarction and AIS,4,11-14 the potential systemic antithrombotic effects of wild-type (wt) ADAMTS13 (rhADAMTS13 marketed as BAX930), and the absence of hemorrhagic risk often observed with rtPA treatment.15
However, wtADAMTS13 is not without its limitations as it requires extremely supra-physiological concentrations, and, like rtPA, its thrombolytic potential is likely to be limited by variable clot composition in AIS patients. Both of these shortcomings can potentially be explained by recent advances in our fundamental understanding of ADAMTS13 function. It is now known that wtADAMTS13 exists in a conformationally quiescent state, in which an autoinhibitory domain-domain interaction limits its proteolytic activity against VWF and maintains its substrate specificity.16,17 This autoinhibitory interaction imposes a closed conformation and is facilitated by 3 flexible linker regions bordering the C-terminal thrombospondin domains.18-20 Initial binding of ADAMTS13 to its substrate, at the VWF D4-CK domains, disrupts this CUB-Spacer interaction so that the open conformation is favored, thereby enhancing activity against VWF. Disruption of the CUB-Spacer interaction can also be achieved through mutation of key residues in the Spacer domain VWF-binding exosites as in the R660K/F592Y/R568K/Y661F/Y665F variant of ADAMTS1321 (also referred to as gain-of-function [GoF] ADAMTS13), which exhibits enhanced activity and broadened substrate specificity.22 In a murine distal middle cerebral artery occlusion (MCAo) model of AIS, the GoF ADAMTS13 variant performed better than wtADAMTS13, partially restoring regional cerebral blood flow (rCBF) to the MCA territory at a lower dose and with a longer therapeutic time window.23 Nevertheless, efficacy of the GoF ADAMTS13 was still limited.
The aim of this study was to design and characterize a panel of novel ADAMTS13 variants, with point mutations in the linker-3 region, with the aim of achieving constitutive ADAMTS13 activity. Our lead variant, Ala1144Val (hereafter referred to as constitutively active [ca] ADAMTS13), was further characterized using in vitro assays of platelet recruitment and substrate specificity before being tested in 2 murine models of AIS.
Materials and methods
In vivo study design
The number of animals required in each treatment group, for both distal MCAo and transient middle cerebral artery occlusion (tMCAo) models, were determined by power calculations based on a 30% reduction in infarct volume (as positive primary outcome), a power of 90%, and a significance level of P < .05.
Animals were assigned to allocation-concealed treatment groups prior to surgery, and the identity of treatment groups remained concealed until all analyses were complete. The criteria to exclude animals in which MCAo failed to produce a 60% reduction in CBF in the ipsilateral hemisphere was set prior to the study.
Animals
All scientific animal procedures were conducted in accordance with the United Kingdom animals (Scientific Procedures) Act (1986) and approved by the local animal welfare and Ethical Review Board of the University of Manchester. Mice were housed in groups (2-5) at 21°C and 65% humidity with a regulated 12-hour light-dark cycle and free access to food and water. Thirty male CD1 mice (Charles River Laboratories) and 18 male C57/BL6 mice (Jackson Laboratories), aged 10 to 13 weeks and weighing 25 to 30 g, were used for distal FeCl3 MCAo and tMCAo, respectively. Twenty-two male CD1 mice were used for tail bleeding assays, and 10 naive male CD1 mice were used as controls for plasma enzyme-linked immunosorbent assays.
Distal FeCl3–mediated MCAo model of stroke
The FeCl3-induced occlusion of the MCA was performed using surgical techniques described previously.4 This model was chosen due to the VWF-dependent thrombotic occlusions formed. Stable occlusion was confirmed by monitoring loss of rCBF in the MCA territory by laser Doppler flow for 1 hour (supplemental Figure 1A, available on the Blood Web site). Laser speckle contrast imaging (see supplemental Methods) was performed at 1-minute intervals for 5 minutes before and 60 minutes after the administration of either vehicle, 6 mg/kg wtADAMTS13, 6 mg/kg caADAMTS13, or 400 mg/kg N-acetyl cysteine (NAC) by tail vein catheter, which was performed at 60 minutes post-FeCl3 application. NAC was included as a partial positive control of MCA recanalisation confirming VWF-dependent thrombosis. Doses were determined by previous studies of ADAMTS1323 and NAC24 in the same model.
Transient MCAo model of ischemia/reperfusion (I/R) injury and platelet aggregated-mediated tissue hypoperfusion
All animals received a 20 ng/kg dose of recombinant mouse IL-1β (R&D Systems) in phosphate-buffered saline containing 0.5% bovine serum albumin by intraperitoneal injection to induce a prestroke systemic inflammatory response before receiving 30 minutes MCAo (see supplemental Methods for surgical procedure) followed by 6 hours reperfusion. At 4 hours of reperfusion (4 hours postfilament withdrawal), animals received 6 mg/kg caADAMTS13 or vehicle by tail vein catheter. Based on a lack of efficacy against platelet aggregates in the distal MCAo study (see “Results”), wtADAMTS13 and NAC were not tested in this model.
Results
The Ala1144Val substitution induces a constitutively active conformation of ADAMTS13
The panel of ADAMTS13 variants was designed to induce the active conformation of the linker-3 region through point mutations in either the alanine-rich “hinge” (Ala1144Val, Ala1145Val, and Ala1146Val, shown in purple in Figure 1A) to reduce conformational flexibility of the region or through point mutations to remove vital “kinks” in the region’s protein backbone (Pro1147Val, Pro1154Val, Pro1171Val, Pro1173Val, Pro1175Val, Pro1180Val, and Pro1182Val, shown in red in Figure 1A). The conformation of these variants was interrogated using an immunoprecipitation assay, in which binding of the cryptic ADAMTS13 spacer domain epitope to the II-1 mAb is indicative of an “open” or active conformation (Figure 1B). In solution, wtADAMTS13 was largely in the “closed” conformation and the majority of the assay input (IN) remained in the flow-through with little being eluted from the antibody-conjugated beads. The proportion of wtADAMTS13 in the “open” conformation, and therefore bound to the beads, was increased upon preincubation with the VWF D4-CK domain fragment. In comparison, the linker-3 variant Ala1144Val was predominantly in the “open” conformation.

Characterization of novel ADAMTS13 variants with constitutive activity. (A) Three-dimensional model of the “open” conformation of ADAMTS13 was constructed using the disintegrin-like domain, thrombospondin type-1 motif, cysteine–rich domain, and spacer domain (DTCS) crystal structure (PDB 3GHM38), homology modeling of the MP-Dis domains, homology modeling of the CUB domains, and the thrombospondin-1 type 1 repeat crystal structure (PDB 1LSL39). Modeling of the linker-3 regions in wtADAMTS13 and Ala1144Val ADAMTS13 was performed using Pymol 2.4. (B) Conformation-dependent immunoprecipitation of ADAMTS13-Myc/His6 proteins. Equal volumes of starting material (IN), the unbound fraction (FT), and immunoprecipitated protein (E) were loaded for comparison. (C) The proteolytic activities of each ADAMTS13 variant were determined using the FRETS-VWF73 assay, performed in the absence (−) and presence (+) of the VWF D4-CK domain fragment. All activities were normalized to that of wtADAMTS13 and are presented as mean plus or minus standard deviation (n = 3-4). (D) The extent of VWF-mediated platelet capture under parallel flow was performed in the presence of increasing concentrations of ADAMTS13 (WT, GoF, or Ala1144Val). Results are presented as mean plus or minus standard deviation (n = 3-5). In each plot, ADAMTS13− (n = 6) and VWF− (n = 3) controls are included for comparison. (E) Fibrinolysis curves were generated by measuring the absorbance of normal human plasma following the addition of thrombin and t-PA, alone or in combination with wtADAMTS13, Ala1144Val ADAMTS13, or NAC. Each timepoint represents the mean ± standard deviation of 6 independent experiments. Individual plots were used to determine lysis times which were compared by ordinary 1-way analysis of variance (ANOVA) with Dunnett’s test of multiple comparisons. *P < .05, ****P < .0001. (F-G) The fibrinolysis assay was adapted to allow thrombin-induced fibrin formation to occur before the addition of t-PA and ADAMTS13. This was performed in a plasma-based assay (F) and purified fibrinogen-based assay (G). Curves are presented as the mean plus or minus standard deviation of 3 independent experiments.
Characterization of novel ADAMTS13 variants with constitutive activity. (A) Three-dimensional model of the “open” conformation of ADAMTS13 was constructed using the disintegrin-like domain, thrombospondin type-1 motif, cysteine–rich domain, and spacer domain (DTCS) crystal structure (PDB 3GHM38), homology modeling of the MP-Dis domains, homology modeling of the CUB domains, and the thrombospondin-1 type 1 repeat crystal structure (PDB 1LSL39). Modeling of the linker-3 regions in wtADAMTS13 and Ala1144Val ADAMTS13 was performed using Pymol 2.4. (B) Conformation-dependent immunoprecipitation of ADAMTS13-Myc/His6 proteins. Equal volumes of starting material (IN), the unbound fraction (FT), and immunoprecipitated protein (E) were loaded for comparison. (C) The proteolytic activities of each ADAMTS13 variant were determined using the FRETS-VWF73 assay, performed in the absence (−) and presence (+) of the VWF D4-CK domain fragment. All activities were normalized to that of wtADAMTS13 and are presented as mean plus or minus standard deviation (n = 3-4). (D) The extent of VWF-mediated platelet capture under parallel flow was performed in the presence of increasing concentrations of ADAMTS13 (WT, GoF, or Ala1144Val). Results are presented as mean plus or minus standard deviation (n = 3-5). In each plot, ADAMTS13− (n = 6) and VWF− (n = 3) controls are included for comparison. (E) Fibrinolysis curves were generated by measuring the absorbance of normal human plasma following the addition of thrombin and t-PA, alone or in combination with wtADAMTS13, Ala1144Val ADAMTS13, or NAC. Each timepoint represents the mean ± standard deviation of 6 independent experiments. Individual plots were used to determine lysis times which were compared by ordinary 1-way analysis of variance (ANOVA) with Dunnett’s test of multiple comparisons. *P < .05, ****P < .0001. (F-G) The fibrinolysis assay was adapted to allow thrombin-induced fibrin formation to occur before the addition of t-PA and ADAMTS13. This was performed in a plasma-based assay (F) and purified fibrinogen-based assay (G). Curves are presented as the mean plus or minus standard deviation of 3 independent experiments.
The conformational change induced by many of these point mutations equates to an approximately four- to fivefold increase in proteolytic activity against the FRETS-VWF73 substrate compared with wtADAMTS13 (Figure 1C). Unlike wtADAMTS13, and similar to the previously reported GoF variant,21,23 their activity was not further enhanced upon addition of the VWF D4-CK domain fragment. To ensure reliability of these activity measurements, the concentration of all ADAMTS13 variants were determined using an appropriate enzyme-linked immunosorbent assay and corroborated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (supplemental Figure 2A-B). The linearity of the FRETS-VWF73 assay at the lower and upper extremes of activity was also confirmed (supplemental Figure 2C-D).
The enhanced activity of caADAMTS13 was also evident in an assay of VWF-mediated platelet capture under arterial shear stress (Figure 1D), in which it completely inhibited platelet capture at subphysiological concentrations (<5 nM). A comparable level of inhibition was not achieved by either WT- or GoF ADAMTS13. The ability of caADAMTS13 to accelerate the proteolysis of fibrin by t-PA/plasmin, a previously identified characteristic of the GoF ADAMTS13 variant,22 was investigated using plasma-based fibrinolysis assays. When present upon initiation of fibrin formation, a significant decrease in 50% lysis time was observed for both wtADAMTS13 (P < .05) and caADAMTS13 (P < .0001) compared with t-PA alone (Figure 1E). The same effect was not observed in the presence of NAC. When overlaid onto preformed fibrin in the plasma-based assay (Figure 1F), caADAMTS13 significantly enhanced the rate of fibrinolysis compared with t-PA alone, reducing the residual absorbance at 120 minutes from 85.3 plus or minus 0.3 to 66.3 plus or minus 4.6. The same effect was not observed with wtADAMTS13. In the same assay performed using purified fibrinogen, t-PA, alone or combined with wtADAMTS13, failed to induce fibrinolysis (Figure 1G). However, the presence of caADAMTS13 induced complete fibrinolysis within 30 minutes.
caADAMTS13 restores rCBF when administered 1 hour after distal MCAo
Contaminating proteins in the wtADAMTS13 and caADAMTS13 preparations used in in vivo experiments (supplemental Figure 2A) were identified by mass spectrometry (supplemental Table 1). The similarity of the contaminants in both preparations and the absence of any proteins with potential anticoagulant, anti-platelet, or thrombolytic activity indicate that all observed in vivo activity is specific to ADAMTS13.
At 1-hour post-FeCl3–induced thrombotic occlusion of the distal MCA (supplemental Figure 1A), we observed a 50% to 60% decrease in mean flux measurements, determined using laser speckle contrast imaging (LSCI), in the region of interest (ROI) encompassing the MCA territory of the ipsilateral hemisphere compared with an equivalent ROI in the contralateral hemisphere (Figure 2A). In vehicle-treated animals, the flux values in both ROIs remained constant over the 1-hour imaging period (Figure 2B), and the ratio of the flux values between the 2 hemispheres remained below the 0.5 threshold assigned to indicate ongoing occlusion of the MCA (Figure 2C). As a result, the mean increase in flux ratio between the time = 0 and time = 60 minutes time points was minimal in the vehicle-treated group (Figure 2E). This prolonged MCAo resulted in large cortical infarcts in vehicle-treated animals (Figure 2D,F). A similarly prolonged occlusion, and resulting lesion, was observed in animals treated with wtADAMTS13.

Restoration of rCBF to the ipsilateral MCA territory following administration of caADAMTS13 at 1 hour post-FeCl3–induced thrombotic MCAo in mice. (A) Representative laser speckle contrast imaging (LCSI) raw data with defined ipsilateral and contralateral ROIs. (B) Flux values from each ROI were extracted at 1-minute intervals for 5 minutes before and 60 minutes after injection. Shown are representative measurements of ipsilateral and contralateral flux from a single animal in each treatment group. (C) Ratios of ipsilateral and contralateral flux values (plus or minus standard deviation, n = 7) are shown for 5 minutes prior to injection, confirming stable occlusion of the MCA as indicated by flux ratios <0.5 (dotted lines). The times after injection at which the flux ratio breached the 0.5 threshold (indicating recanalisation) are designated by an asterisk. For comparison, the flux ratio from a single animal undergoing sham surgery is also shown (top panel). (D) Representative cresyl violet–stained coronal sections from a single animal in each treatment group used in the determination of cortical lesion volumes. Scale bars, 500 μm. (E) Percentage increase in flux ratio between 0 and 60 minutes postinjection determined for each individual animal; error bars represent mean ± standard deviation. (F) Lesion volumes were determined at 24 hours postocclusion by measuring the area of cell death over 3 to 9 coronal levels; error bars represent mean ± standard deviation. Single comparisons were performed using an unpaired Student t test. **P < .01.
Restoration of rCBF to the ipsilateral MCA territory following administration of caADAMTS13 at 1 hour post-FeCl3–induced thrombotic MCAo in mice. (A) Representative laser speckle contrast imaging (LCSI) raw data with defined ipsilateral and contralateral ROIs. (B) Flux values from each ROI were extracted at 1-minute intervals for 5 minutes before and 60 minutes after injection. Shown are representative measurements of ipsilateral and contralateral flux from a single animal in each treatment group. (C) Ratios of ipsilateral and contralateral flux values (plus or minus standard deviation, n = 7) are shown for 5 minutes prior to injection, confirming stable occlusion of the MCA as indicated by flux ratios <0.5 (dotted lines). The times after injection at which the flux ratio breached the 0.5 threshold (indicating recanalisation) are designated by an asterisk. For comparison, the flux ratio from a single animal undergoing sham surgery is also shown (top panel). (D) Representative cresyl violet–stained coronal sections from a single animal in each treatment group used in the determination of cortical lesion volumes. Scale bars, 500 μm. (E) Percentage increase in flux ratio between 0 and 60 minutes postinjection determined for each individual animal; error bars represent mean ± standard deviation. (F) Lesion volumes were determined at 24 hours postocclusion by measuring the area of cell death over 3 to 9 coronal levels; error bars represent mean ± standard deviation. Single comparisons were performed using an unpaired Student t test. **P < .01.
In animals treated with NAC or caADAMTS13, we observed a gradual restoration of CBF in the ipsilateral ROI (Figure 2A-B). This effect was more pronounced in the caADAMTS13-treated group, in which the flux ratio breached the 0.5 threshold (denoted by an asterisk on Figure 2C and indicating MCA recanalisation) at an earlier time point than in the NAC-treated group (24 minutes and 44 minutes, respectively). When compared with the vehicle-treated group, a significant increase in flux ratio and accompanying decrease in lesion volume was only observed in the caADAMTS13-treated group (Figure 2E-F).
An important observation made throughout the course of this study was the absence of hemorrhagic transformation in any of the treatment groups. Although some small bleeds were apparent within the infarct lesion in some animals (supplemental Figure 3A), their size or frequency did not appear to be associated with any particular treatment group (supplemental Figure 3B). To confirm that there was no hemostatic deficit associated with the treatments, we performed tail bleeding assays and observed no significant alteration of tail bleeding times (supplemental Figure 3C). In plasma samples taken at 24 hours post-MCAo, neither the concentration and collagen binding activity of VWF nor the concentration of fibrinogen was significantly altered in any of the treatment groups compared with vehicle-treated and naive control animals (supplemental Figure 3D-F).
caADAMTS13 reduces the formation of platelet aggregation in the cerebral microvasculature after distal MCAo
At 24 hours postocclusion, there was extensive thrombotic material, positive for both fibrin and VWF, remaining in the MCA, other pial vessels, and penetrating vessels in vehicle-treated animals (Figure 3A). VWF staining colocalized with CD41+ particles, indicative of VWF-platelet aggregate formation (Figure 3E), and this was particularly prominent in the small penetrating arterioles downstream of the main thrombus site where we observed extensive endothelin-1 (ET-1) staining (Figure 3F). Expression of ET-1 significantly correlated with both VWF deposition and the formation of platelet aggregates (supplemental Figure 4A-B).

Potent thrombolytic and antiplatelet activity of caADAMTS13 following distal thrombotic MCAo in mice. (A) The extent of VWF and fibrin(ogen) deposition at the site of the MCAo (#) and in the downstream vessels (white arrows) was visualized by immunofluorescence (IF). (B-C) Thresholded single-channel images were used to quantify the total area of VWF and fibrin(ogen)+ staining, which was normalized for background fluorescence using an equivalent area of the contralateral hemisphere. (D) The number of CD41+ particles, with an area greater than 0.2 μm2, was measured using automated particle counting applied to thresholded images in ImageJ. (E) Platelet aggregates (positive for both VWF and CD41) in the penetrating arterioles are indicated by white arrows. (F-G) The extent of ET-1 staining in the pial vessels was visualized by IF (white arrows) and was quantified/normalized using the same procedure. All images are a representative field of view, chosen to visualize the main site of thrombosis, isolated from whole-brain imaging performed using Slidescanner. Scale bars, 100 μm. Error bars represent mean ± standard deviation. Single comparisons were performed using an unpaired Student t test. *P < .05, **P < .01.
Potent thrombolytic and antiplatelet activity of caADAMTS13 following distal thrombotic MCAo in mice. (A) The extent of VWF and fibrin(ogen) deposition at the site of the MCAo (#) and in the downstream vessels (white arrows) was visualized by immunofluorescence (IF). (B-C) Thresholded single-channel images were used to quantify the total area of VWF and fibrin(ogen)+ staining, which was normalized for background fluorescence using an equivalent area of the contralateral hemisphere. (D) The number of CD41+ particles, with an area greater than 0.2 μm2, was measured using automated particle counting applied to thresholded images in ImageJ. (E) Platelet aggregates (positive for both VWF and CD41) in the penetrating arterioles are indicated by white arrows. (F-G) The extent of ET-1 staining in the pial vessels was visualized by IF (white arrows) and was quantified/normalized using the same procedure. All images are a representative field of view, chosen to visualize the main site of thrombosis, isolated from whole-brain imaging performed using Slidescanner. Scale bars, 100 μm. Error bars represent mean ± standard deviation. Single comparisons were performed using an unpaired Student t test. *P < .05, **P < .01.
Quantification of all of these immunofluorescence markers identified a significant decrease in VWF deposition (Figure 3B), fibrin deposition (Figure 3C), platelet aggregates (Figure 3D), and ET-1 release (Figure 3G) in the caADAMTS13-treated group compared with vehicle-treated. The same effect was not observed in either the NAC- or wtADAMTS13-treated groups.
caADAMTS13 reduces neutrophil migration into ischemic tissue following distal MCAo
In vehicle-treated animals, at 24 hours postocclusion, we observed widespread infiltration of neutrophils into the ischemic tissue (Figure 4A-B). Neutrophils (Ly-6G+ and 4′,6-diamidino-2-phenylindole–stained particles) were most often identified in VWF-stained penetrating arterioles downstream of the main thrombus but could also be seen transmigrating across the vessel wall and were present in the parenchyma, where they appeared to be distinct from any visible vessels (Figure 4C). Neutrophil recruitment correlated significantly with VWF staining, microglial activation, and lesion volume (supplemental Figure 4C-E). At the border of the ischemic tissue, we observed a characteristic activation of microglia, indicated by altered morphology compared with microglia in the contralateral hemisphere (Figure 4D-E). The number of activated microglia correlated significantly with lesion volume (supplemental Figure 4F). Both neutrophil recruitment and microglial activation were significantly reduced in caADAMTS13-treated animals to the point that they were similar to sham animals.
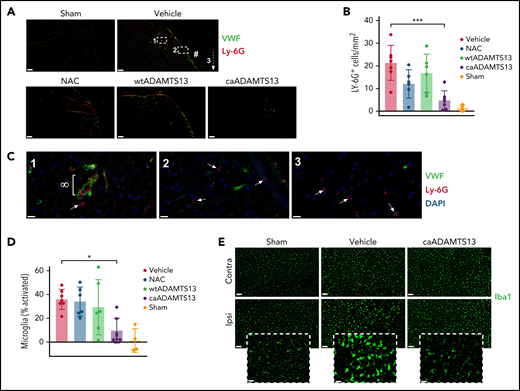
Mice treated with caADAMTS13 1 hour after distal thrombotic MCAo exhibit reduced neutrophil recruitment to the brain parenchyma and reduced peri-infarct microglial activation at 24 hours postocclusion. (A) Recruitment of neutrophils to the ischemic tissue was visualized by IF staining for the neutrophil marker Ly-6G. Images are a representative field of view, chosen to visualize the main site of thrombosis (#), isolated from whole-brain imaging performed using Slidescanner. Scale bars, 200 μm. (B) The number of Ly-6G+ particles, with an area greater than 80 μm2, was measured using automated particle counting applied to thresholded images in ImageJ. (C) Higher magnification images of regions 1 to 3 (indicated in panel A) of a brain section from a vehicle-treated animal. Neutrophils recruited to a VWF-lined vessel (∞) and 4′,6-diamidino-2-phenylindole/Ly-6G–stained cells (presumed to be neutrophils) in the brain parenchyma (white arrows) were observed. Scale bars, 20 μm. (D) The total number of Iba1+ cells and the number of Iba1+ cells with a circularity >0.5 were counted by automated particle analysis of thresholded single-channel images and used to quantify the percentage of total microglia with an activated morphology. (E) Representative Iba1 images from the peri-infarct cortical region of the ipsilateral hemisphere and a corresponding region of the contralateral hemisphere for comparison. Scale bars, 100 μm. Inset images are the ipsilateral field of view at higher magnification to show microglial morphology used to determine activation status. Scale bars, 50 μm. Error bars represent mean ± standard deviation. Single comparisons were performed using an unpaired Student t test. *P < .05, ***P < .001.
Mice treated with caADAMTS13 1 hour after distal thrombotic MCAo exhibit reduced neutrophil recruitment to the brain parenchyma and reduced peri-infarct microglial activation at 24 hours postocclusion. (A) Recruitment of neutrophils to the ischemic tissue was visualized by IF staining for the neutrophil marker Ly-6G. Images are a representative field of view, chosen to visualize the main site of thrombosis (#), isolated from whole-brain imaging performed using Slidescanner. Scale bars, 200 μm. (B) The number of Ly-6G+ particles, with an area greater than 80 μm2, was measured using automated particle counting applied to thresholded images in ImageJ. (C) Higher magnification images of regions 1 to 3 (indicated in panel A) of a brain section from a vehicle-treated animal. Neutrophils recruited to a VWF-lined vessel (∞) and 4′,6-diamidino-2-phenylindole/Ly-6G–stained cells (presumed to be neutrophils) in the brain parenchyma (white arrows) were observed. Scale bars, 20 μm. (D) The total number of Iba1+ cells and the number of Iba1+ cells with a circularity >0.5 were counted by automated particle analysis of thresholded single-channel images and used to quantify the percentage of total microglia with an activated morphology. (E) Representative Iba1 images from the peri-infarct cortical region of the ipsilateral hemisphere and a corresponding region of the contralateral hemisphere for comparison. Scale bars, 100 μm. Inset images are the ipsilateral field of view at higher magnification to show microglial morphology used to determine activation status. Scale bars, 50 μm. Error bars represent mean ± standard deviation. Single comparisons were performed using an unpaired Student t test. *P < .05, ***P < .001.
Restoration of rCBF still occurs when caADAMTS13 is administered 4 hours after proximal tMCAo
We also examined the efficacy of delayed caADAMTS13 treatment in a second disease model in which MCAo is transient and performed in the presence of a systemic inflammatory stimulus (supplemental Figure 1B). The resulting ischemia/reperfusion injury and immediate inflammatory response induces microvascular platelet aggregate formation and tissue hypoperfusion within 4 hours of MCAo and before caADAMTS13 administration. In all animals, the ratio of flux values between the contralateral and ipsilateral ROIs was reduced by 50% to 60% (between baseline and time = 0 images) upon filament insertion (Figure 5A-B). Once the filament was removed and perfusion of the MCA was reestablished, we observed a gradual restoration of tissue perfusion until 4 hours postocclusion (Figure 5B-C). At this point, vehicle-treated animals exhibited a loss of tissue perfusion, between 4 and 6 hours postocclusion, that was significantly greater than that observed in animals treated with caADAMTS13. In vehicle-treated animals, tissue hypoperfusion was accompanied by the development of large striatal lesions (Figure 5D) and extensive IgG leakage (Figure 5E). Both lesion volume and the volume of IgG staining were significantly reduced in caADAMTS13-treated animals. Little evidence of hemorrhagic transformation was identified in either treatment group (supplemental Figure 3G).

Delayed administration of caADAMTS13 restores CBF, reduces infarct volume, and attenuates IgG leakage at 6 hours post-I/R injury in mice. (A) Representative LCSI raw data with defined ipsilateral and contralateral ROIs. (B) Ratios of ipsilateral and contralateral flux values (mean ± standard deviation, n = 8-10) are shown for each time point confirming transient occlusion of the MCA at time = 0 as indicated by flux ratios <0.5 in both treatment groups. (C) Flux ratios were normalized to that of the baseline measurement (dotted line). Error bars represent mean ± standard deviation. Divergence of flux ratios between the 2 treatment groups at each imaging time point was assessed using 2-way ANOVA followed by Sidak’s multiple comparisons test. *P < .05, **P < .01. (D-E) Infarct volume and IgG staining volume, respectively, were determined from whole-brain imaging performed using Slidescanner. Shown are representative images, at an approximately equivalent coronal level, for 2 animals in both treatment groups. Single comparisons were performed using an unpaired Student t test. *P < .05.
Delayed administration of caADAMTS13 restores CBF, reduces infarct volume, and attenuates IgG leakage at 6 hours post-I/R injury in mice. (A) Representative LCSI raw data with defined ipsilateral and contralateral ROIs. (B) Ratios of ipsilateral and contralateral flux values (mean ± standard deviation, n = 8-10) are shown for each time point confirming transient occlusion of the MCA at time = 0 as indicated by flux ratios <0.5 in both treatment groups. (C) Flux ratios were normalized to that of the baseline measurement (dotted line). Error bars represent mean ± standard deviation. Divergence of flux ratios between the 2 treatment groups at each imaging time point was assessed using 2-way ANOVA followed by Sidak’s multiple comparisons test. *P < .05, **P < .01. (D-E) Infarct volume and IgG staining volume, respectively, were determined from whole-brain imaging performed using Slidescanner. Shown are representative images, at an approximately equivalent coronal level, for 2 animals in both treatment groups. Single comparisons were performed using an unpaired Student t test. *P < .05.
caADAMTS13 induces dissolution of platelet aggregates formed early after tMCAo
Ischemia/reperfusion injury, when preceded by an acute systemic inflammatory stimulus, is known to induce platelet aggregate formation in cerebral capillaries (the proposed cause of microvascular reperfusion deficits) as early as 3 hours postocclusion.25 In vehicle-treated animals, we observed significantly higher numbers of platelet aggregates, particularly those with an area 10 to 30 μm2 and >30 μm2, in the ipsilateral striatum compared with that of the contralateral hemisphere (Figure 6A,C). A smaller and not statistically significant increase in platelet aggregates was also observed in the ipsilateral cortex compared with the contralateral cortex. In contrast, there was no significant difference in the number of platelet aggregates, of any size, in either the cortex or striatum of the ipsilateral hemisphere, compared with the contralateral hemisphere, in caADAMTS13-treated animals (Figure 6B-C).
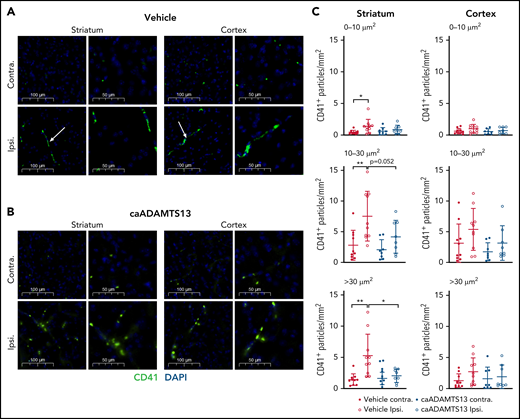
caADAMTS13-mediated dissolution of preformed platelet aggregates in the mouse brain following tMCAo and I/R injury. (A-B) Visualization of platelet aggregates (white arrows) in vehicle and caADAMTS13-treated animals, respectively, was achieved by IF staining for the constitutive platelet marker CD41. Images are representative fields of view from either the striatum or cortex of the ipsilateral hemisphere and equivalent regions of the contralateral hemisphere. Each field of view is shown at 2 magnifications. Scale bars, 100 or 50 μm. (C) Thresholded single channel images were used for automatic particle counting in ImageJ, selecting aggregates based on particle size (0-10, 10-30, and >30 μm2). Error bars represent mean plus or minus standard deviation. Single comparisons were performed using an unpaired Student t test. *P < .05, **P < .01.
caADAMTS13-mediated dissolution of preformed platelet aggregates in the mouse brain following tMCAo and I/R injury. (A-B) Visualization of platelet aggregates (white arrows) in vehicle and caADAMTS13-treated animals, respectively, was achieved by IF staining for the constitutive platelet marker CD41. Images are representative fields of view from either the striatum or cortex of the ipsilateral hemisphere and equivalent regions of the contralateral hemisphere. Each field of view is shown at 2 magnifications. Scale bars, 100 or 50 μm. (C) Thresholded single channel images were used for automatic particle counting in ImageJ, selecting aggregates based on particle size (0-10, 10-30, and >30 μm2). Error bars represent mean plus or minus standard deviation. Single comparisons were performed using an unpaired Student t test. *P < .05, **P < .01.
In this model, we observed extensive microglial activation in vehicle-treated animals at 6 hours postocclusion, indicated by a significant increase in the number of microglia with an activated morphology and an increase in the number of IL-1α+ microglia, in the peri-infarct region of the ipsilateral hemisphere (Figure 7A,C). Significantly increased numbers of IL-1α+ microglia were also observed in the remainder of the striatum and in the cortex of the ipsilateral hemisphere compared with the contralateral (Figure 7C). Treatment with caADAMTS13 at 4 hours postocclusion had little impact on the number of microglia exhibiting an activated morphology in the peri-infarct region; however, we did observe a significant reduction in the number of microglia expressing IL-1α compared with those in the vehicle-treated group (Figure 7B-C).
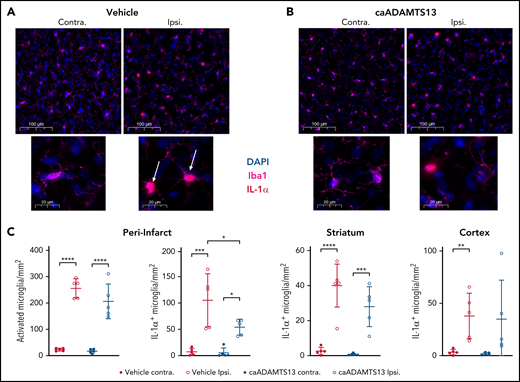
Delayed caADAMTS13 administration after tMCAo and I/R injury in mice reduces IL-1α expression by peri-infarct microglia. (A-B) Microglia in the brains of vehicle- and caADAMTS13-treated animals, respectively, were stained by IF for both Iba1 and IL-1α. Images are representative fields of view from the peri-infarct region of the ipsilateral hemisphere and an equivalent region of the contralateral hemisphere. Each field of view is shown at 2 magnifications. Scale bars, 100 or 20 μm. (C) These images, and images taken from the striatum and cortex of both hemispheres, were analyzed by automated particle counting to identify activated microglia, based on morphology in Iba1 staining, or IL-1α–expressing microglia. Error bars represent mean ± standard deviation. Single comparisons were performed using an unpaired Student t test. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Delayed caADAMTS13 administration after tMCAo and I/R injury in mice reduces IL-1α expression by peri-infarct microglia. (A-B) Microglia in the brains of vehicle- and caADAMTS13-treated animals, respectively, were stained by IF for both Iba1 and IL-1α. Images are representative fields of view from the peri-infarct region of the ipsilateral hemisphere and an equivalent region of the contralateral hemisphere. Each field of view is shown at 2 magnifications. Scale bars, 100 or 20 μm. (C) These images, and images taken from the striatum and cortex of both hemispheres, were analyzed by automated particle counting to identify activated microglia, based on morphology in Iba1 staining, or IL-1α–expressing microglia. Error bars represent mean ± standard deviation. Single comparisons were performed using an unpaired Student t test. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Compared with the FeCl3 MCAo model, in which ex vivo outputs are measured at 24 hours postocclusion, this early pathology appears to involve less VWF-mediated neutrophil recruitment, as might be expected given that the animals are culled at 6 hours postocclusion. As such, the protective effect of caADAMTS13 in this respect is much less pronounced (supplemental Figure 5).
Discussion
The therapeutic landscape for the treatment of AIS has looked bleak for some time. Despite promising preclinical and early clinical trials, few candidate pharmacological therapies have fulfilled their early promise, and none have been successful in replacing rtPA (or its derivative Tenecteplase26) as the first line hyperacute thrombolytic therapy. The ideal candidate, in addition to thrombolytic potential against thrombi with differing compositions, should also address the immune and inflammatory elements of stroke pathology, which can prove so detrimental to outcome even when CBF is effectively restored.27 Together with the absence of any hemorrhagic risk and a longer therapeutic time window to facilitate adequate uptake of the treatment, these are a challenging set of criteria. The overarching aim of this study was to build upon the recent developments in our understanding of ADAMTS13 function, and the partially successful preclinical studies of wtADAMTS13 and GoF ADAMTS13, to develop a novel ADAMTS13-based thrombolytic with an activity profile that could fulfill these criteria in the preclinical setting.
By examining the sequence of the ADAMTS13 linker-3 region, postulated by Deforche et al to be one of the structural features required for conformational activation,20 we identified a patch of alanine residues (Ala1144, Ala1145, and Ala1146). This appeared to represent a molecular “hinge” that would facilitate the conformational change required for activation of ADAMTS13. Point mutation of 2 of these alanine residues (Ala1144 and Ala1146) to valine, to reduce their innate flexibility, or mutation of 2 out of the 7 proline residues (Pro1180 and Pro1182) to valine, to alter the region’s resting structure, instilled an apparent constitutive activity. The activity of these variants is consistently enhanced by fivefold, compared with wtADAMTS13, and is higher than observed previously in the GoF variant,16 suggesting the variants are locked in an open conformation as opposed to substrate-activated wtADAMTS13 or GoF ADAMTS13, which preferentially adopt the open form. This assumption is further indicated by the results of conformation-dependent immunoprecipitation assays performed on the lead variant Ala1144Val.
The further enhancement of activity against platelet capture in caADAMTS13 and a similar alteration of substrate specificity to that observed previously in GoF ADAMTS1322 suggested potential for markedly enhanced systemic thrombolytic activity in vivo compared with either wtADAMTS13 or GoF ADAMTS13.4,23 In this study, we performed a direct comparison of caADAMTS13 against wtADAMTS13 and NAC in the distal MCAo model under our experimental conditions. These 2 treatments have both been tested previously by other groups,4,24 although only rCBF and lesion volume were measured, and, in the case of NAC, administration was performed at only 20 minutes postocclusion. Although both wtADAMTS13 and NAC exerted some level of improvement (compared with the vehicle-treated group) in 1 or 2 of the key measured outputs, only caADAMTS13 achieved significant improvement in all outputs including restoration of rCBF and reduction of lesion volume (P < .01 in both cases). Failure of wtADAMTS13 to restore CBF, at the dose and time of delayed administration used in this study, has been reported previously23 and is unsurprising given the requirement for substrate-induced activation.
There are numerous specific properties of caADAMTS13 that are attractive for potential translation as a thrombolytic therapy. Of these, perhaps the most important is its proteolytic activity against both VWF and fibrinogen. As suggested by in vitro experiments, caADAMTS13 is capable of clearing both VWF and fibrin deposits from the thrombus. This indicates that its efficacy for treating AIS in clinic will be less dependent on clot composition.3,4 It is currently unknown whether caADAMTS13 possesses proteolytic activity against other substrates, which could be contributing to the protective effects reported here and which should be considered in future studies of off-target effects.
The potent proteolytic activity of caADAMTS13 against VWF in in vitro assays translates to an almost complete degradation of VWF in all vessels within the ischemic tissue. This appears to influence the ability of VWF to recruit leukocytes. A reduction in the number of neutrophils recruited to the ischemic tissue was exclusively observed in the caADAMTS13 treatment group, and this is likely to dampen downstream inflammatory processes (including microglial activation), thereby protecting tissue from the detrimental effects of aberrant inflammation. This undoubtedly contributes to the reduction in lesion volume observed here (at 24 hours poststroke) but may also reduce progression of the stroke lesion in the acute and subacute phases through reduction of monocyte/macrophage infiltration.8 Degradation of VWF at the site of injury may also influence leukocyte recruitment indirectly through a reduction in platelet adhesion. The direct action of caADAMTS13 on VWF-dependent neutrophil recruitment is currently under investigation.
In this study, caADAMTS13 was examined in 2 different models of murine stroke. Having observed a reduction in the number of platelet aggregates in the penetrating vessels following distal MCAo in the caADAMTS13 treatment group, we introduced another experimental protocol to confirm that this was due to active degradation of these aggregates and not simply the inhibition of their formation (as caADAMTS13 was administered at 1 hour poststroke). In this protocol, tMCAo with I/R injury was performed in the presence of systemic inflammation to induce early formation of platelet aggregates and the so-called “no reflow” phenomenon in which microvascular platelet plugging interferes with tissue reperfusion despite recanalisation of the MCA.25 A 4-hour delay in the administration of caADAMTS13 allowed for preformation of these aggregates and confirmed their active degradation. Importantly, this experiment not only confirmed a positive effect of caADAMTS13 on tissue no-reflow but also an extended therapeutic time window in general, as CBF restoration, lesion volume reduction, and reduced microglial activation were still observed.
This persistent efficacy, despite delayed administration, is another property of caADAMTS13 that could potentially facilitate effective translation of the therapy to AIS patients as a narrow therapeutic time window is a common translational road-block in many countries. This issue has also limited the effective application of rtPA to AIS patients, which, despite efforts to extend the therapeutic window,30 is hindered by a reduction in efficacy and significant risk of hemorrhagic transformation when administered beyond 3 to 4.5 hours poststroke.29,30 Recent evidence suggests this can be extended to 4.5 to 9 hours poststroke when perfusion-diffusion magnetic resonance imaging or computed tomography perfusion imaging is available,31 although this is a resource that is not widely available. In both murine stroke models used here, including that in which caADAMTS13 is administered at 4 hours poststroke, we have observed no evidence of any hemorrhagic transformation or any systemic reduction of VWF or fibrinogen. In fact, there is a suggestion that caADAMTS13 might reduce the incidence of hemorrhage associated with MCAo, presumably through its action on leukocyte recruitment and protection against blood brain barrier breakdown as indicated by a reduction in IgG leakage. This may even also suggest a potential protective effect of caADAMTS13 in subarachnoid and/or intracerebral hemorrhage as has been observed previously with rhADAMTS13.32,33 This would further improve the potential application of the therapy to AIS patients, removing the need for confirmatory imaging prior to administration and hence much more rapid delivery of treatment.
Further preclinical studies to confirm the absence of off-target effects and to confirm caADAMTS13’s mode of action are required before the commencement of phase 1 dose-escalation trials to evaluate efficacy and safety in human participants. The thrombolytic potential of caADAMTS13, although a very exciting development in the pursuit of novel stroke therapies, may also be applicable to a number of thrombotic and inflammatory pathologies involving VWF. This includes, but is not limited to, myocardial infarction,34 TTP,35 chronic thromboembolic pulmonary hypertension,36 and inflammatory bowel disease.37
Acknowledgments
This work was funded by a British Heart Foundation project grant (PG/17/86/33399), an MRC research grant (MR/N003586/1), a Medical Research Foundation fellowship (MRF-076-0004-RG-SOUT-C0756 [K.S.]), and a PhD studentship from King Abdulaziz University, Jeddah, Saudi Arabia (KAU1763 [O.S.]).
Authorship
Contribution: K.S., O.S., E.L., and G.C. performed the data collection; K.S. and O.S. generated the figures and analyzed the data; K.S., I.S., C.J.S., and S.M.A. designed the study; and K.S., O.S., I.S., C.J.S., and S.M.A. interpreted the data and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Kieron South, Room 2.012, AV Hill Building, University of Manchester, Oxford Rd, Manchester M13 9PT, United Kingdom; e-mail: kieron.south@manchester.ac.uk.
All data are available by request to the corresponding author via e-mail. The Ala1144Val ADAMTS13 variant and related linker-3 variants are the subject of GB patent application 2102208.2 (“A novel biologic for treatment of thrombotic indications”), which has been filed but will not be in the public domain until 17 August 2022.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal