


Abstract
Disseminated intravascular coagulation (DIC) is a syndrome triggered by infectious and noninfectious pathologies characterized by excessive generation of thrombin within the vasculature and widespread proteolytic conversion of fibrinogen. Despite diverse clinical manifestations ranging from thrombo-occlusive damage to bleeding diathesis, DIC etiology commonly involves excessive activation of blood coagulation and overlapping dysregulation of anticoagulants and fibrinolysis. Initiation of blood coagulation follows intravascular expression of tissue factor or activation of the contact pathway in response to pathogen-associated or host-derived, damage-associated molecular patterns. The process is further amplified through inflammatory and immunothrombotic mechanisms. Consumption of anticoagulants and disruption of endothelial homeostasis lower the regulatory control and disseminate microvascular thrombosis. Clinical DIC development in patients is associated with worsening morbidities and increased mortality, regardless of the underlying pathology; therefore, timely recognition of DIC is critical for reducing the pathologic burden. Due to the diversity of triggers and pathogenic mechanisms leading to DIC, diagnosis is based on algorithms that quantify hemostatic imbalance, thrombocytopenia, and fibrinogen conversion. Because current diagnosis primarily assesses overt consumptive coagulopathies, there is a critical need for better recognition of nonovert DIC and/or pre-DIC states. Therapeutic strategies for patients with DIC involve resolution of the eliciting triggers and supportive care for the hemostatic imbalance. Despite medical care, mortality in patients with DIC remains high, and new strategies, tailored to the underlying pathologic mechanisms, are needed.
Introduction
Disseminated intravascular coagulation (DIC) is a life-threatening thrombohemorrhagic condition triggered by infectious and noninfectious causes. According to the International Society for Thrombosis and Hemostasis (ISTH),1 DIC is an acquired syndrome characterized by systemic activation of coagulation within the vasculature leading to microvascular damage and organ dysfunction. The definition emphasizes that DIC originates, develops within, and affects the microvasculature and is not restricted to the site of insult, but spreads throughout the body.
Etiology
DIC is a complication of many disorders, such as severe systemic infections, malignancy, trauma, obstetrical complications, vascular malformations, severe immunological reactions, and heat stroke. Despite a heterogenous clinical presentation ranging from asymptomatic, to mild, to overwhelming thrombosis or bleeding, at its core, DIC follows exposure to or production of procoagulants insufficiently balanced by endogenous anticoagulant and fibrinolytic mechanisms. The excessive activation of thrombin, evidenced by elevated thrombin-antithrombin complexes or prothrombin activation fragments in the circulation, leads to proteolytic fibrinogen conversion and intravascular formation of fibrin. If usage of clotting factors exceeds the synthetic hepatic output, a consumptive coagulopathy develops that, in conjunction with thrombocytopenia, predicts an elevated risk of bleeding. Intravascular fibrin is counterbalanced by plasmin-mediated fibrinolysis leading to increased fibrin degradation products (D-dimers) in the circulation. When fibrinolysis cannot compensate for the excessive coagulopathy, obstructive microthrombi formation may lead to hypoperfusion and hypoxia of peripheral organs.
Clinical features
Signs and symptoms of DIC include bleeding, bruising, low blood pressure, shortness of breath, and confusion. Clinical manifestations comprise (1) microvascular thrombosis that leads to extensive organ dysfunction, gangrenes, acute kidney injury, and sometimes pulmonary and cerebral thrombosis; (2) bleeding such as petechiae, ecchymoses and necrotizing purpura in the skin; bleeding from vascular access sites; and mucosal bleeding into the gastrointestinal tract, pulmonary alveolae, adrenals, and central nervous system; and (3) shock due to blood loss, impaired endothelial permeability, and/or decreased vascular tone.
Microvascular thrombosis and bleeding are the major causes of multiple organ failures, with lungs and kidneys being predominantly sensitive to coagulopathic dysfunction. Pulmonary thrombosis impairs gas exchanges, leading to hypoxemia, and damages the alveolar blood-air barrier, resulting in flooding of the alveolae with plasma (edema) and blood cells (hemorrhages), which contribute to acute respiratory distress syndrome. Likewise, glomerular and peritubular microthrombosis causes hypoperfusion and subsequent ischemia-reperfusion damage, impairs urinary filtration, and induces acute kidney injury.
Epidemiology
The incidence of DIC is variable according to geographic location, clinical setting, underlying condition, and diagnostic criteria.2 DIC incidence in various medical conditions is depicted in Figure 1. Mortality rates range from 31% to 86% despite supportive therapy, and are always higher than in patients without DIC. The presence of DIC is the strongest predictor of death within 28 days in patients with sepsis.2
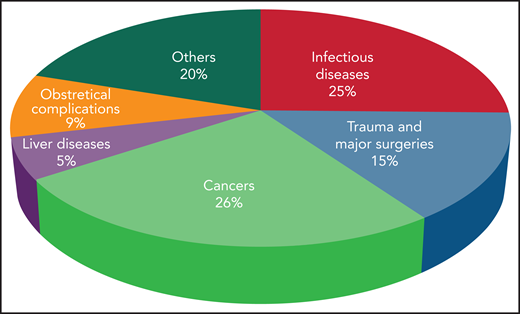
Incidence of DIC in critically ill patients grouped according to the main underlying disease.
Incidence of DIC in critically ill patients grouped according to the main underlying disease.
Diagnosis
Considering the multitude of causative factors, DIC diagnosis is always made in the context of the clinical presentation. Currently, DIC cannot be diagnosed by any single laboratory test, and combinations of clinical biomarkers are used instead. Global coagulation tests, such as activated partial thromboplastin time and prothrombin time (PT) are routinely used in patients with sepsis to assess the consumptive coagulopathy. Although not yet validated for diagnostic use, viscoelastic point-of-care testing allows for rapid and concomitant monitoring of both coagulation and fibrinolysis in patients3 treated for trauma and, in pilot studies,4 distinguished between critically ill patients with DIC and patients without DIC.
Three diagnostic algorithms for DIC have been developed, 2 in Japan and 1 by the ISTH Standardization Committee.5 The ISTH diagnostic score for overt DIC (Table 1) is calculated based on platelet count, PT, fibrinogen level, and fibrinogen/fibrin degradation peptides (D-dimer) in conjunction with clinical considerations.1 A score of 5 or more indicates overt DIC. A simpler DIC score calculated in the first 48 hours using platelet counts and PT could reflect the severity of the underlying disorder.6
ISTH diagnostic algorithm for the diagnosis of overt DIC
| Parameter/result | Score* |
|---|---|
| Underlying disease | |
| No | — |
| Yes; proceed | — |
| Platelet count | |
| >100 ×109/L | 0 |
| <100 ×109/L but >50 ×109/L | 1 |
| <50 ×109/L | 2 |
| Prolonged PT | |
| <3 s | 0 |
| >3 s but <6 s | 1 |
| >6 s | 2 |
| Elevated fibrin-related marker (eg, soluble fibrin monomer/fibrin degradation products) | |
| No increase | 0 |
| Moderate increase | 2 |
| Strong increase | 3 |
| Fibrinogen level | |
| >1.0 g/L | 0 |
| <1.0 g/L | 1 |
| Parameter/result | Score* |
|---|---|
| Underlying disease | |
| No | — |
| Yes; proceed | — |
| Platelet count | |
| >100 ×109/L | 0 |
| <100 ×109/L but >50 ×109/L | 1 |
| <50 ×109/L | 2 |
| Prolonged PT | |
| <3 s | 0 |
| >3 s but <6 s | 1 |
| >6 s | 2 |
| Elevated fibrin-related marker (eg, soluble fibrin monomer/fibrin degradation products) | |
| No increase | 0 |
| Moderate increase | 2 |
| Strong increase | 3 |
| Fibrinogen level | |
| >1.0 g/L | 0 |
| <1.0 g/L | 1 |
Adapted from Taylor et al.1
If ≥5, compatible with overt DIC, repeat scoring daily; if <5, suggestive (not affirmative) of nonovert DIC, repeat next in 1 to 2 d.
There have been attempts to establish scoring algorithms to detect early (nonovert, compensated) DIC before it reaches a full-blown, frequently irreversible stage.1,7 Biomarkers of thrombin generation (TAT and prothrombin fragment-1 and -2), fibrinolysis activation (plasmin-antiplasmin and D-dimer), and suppression (plasminogen activator inhibitor 1 [PAI1]; tissue plasminogen activator [t-PA]-PAI-1 complexes) are used to assess early stages of DIC, as well as its severity and progression. Measurement of damage-associated molecular patterns (DAMPs), such as high-mobility group box-1, nucleosomes, extracellular histones, and cell-free DNA, in correlation with scores of organ function (Sequential Organ Failure Assessment and Multiple Organ Dysfunction Score) and severity of disease (Acute Physiology and Chronic Health Evaluation II), could provide additional information, but their diagnostic and prognostic potential has not been demonstrated in large cohorts. Currently, the absence of algorithms with DIC prediction power greatly limits preventive therapeutic interventions.
Pathogenesis of DIC
As summarized in Figure 2, DIC pathophysiology is complex and multifactorial and involves overlapping host defense pathways, such as uncontrolled activation of coagulation, platelets, fibrinolysis, complement, innate immunity, and inflammation, within a dysfunctional microcirculation, characterized by widespread endotheliopathy.
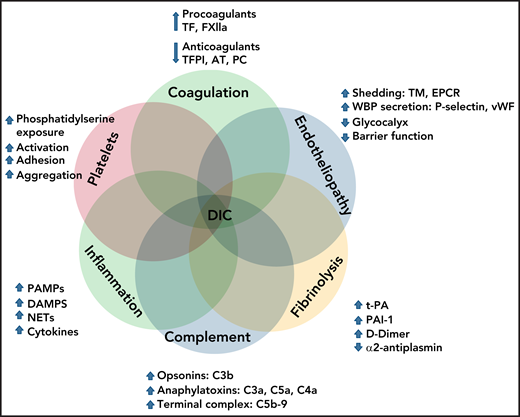
Interactions of cellular and molecular components of host defense in the pathogenesis of DIC.
Interactions of cellular and molecular components of host defense in the pathogenesis of DIC.
Initiation of coagulation
Blood coagulation is primarily triggered by the exposure of tissue factor (TF) to coagulation factor (F) VII/VIIa8 circulating in the plasma. In sepsis and likely other pathologies, activation of the contact pathway can also contribute to the initiation and amplification of the coagulopathic and inflammatory responses.9,10 TF is constitutively expressed outside vasculature where it forms a protective hemostatic envelope, but is normally repressed on vascular cells. Inducible TF expression in monocytes,11,12 platelets,13,14 granulocytes,15,16 lymphocytes,17 and endothelial cells18,19 has been reported. Monocytes are considered the primary source of intravascular TF in DIC, whereas TF transcription in other vascular cells has been challenged.20 Multiple immune signaling networks promote intravascular TF expression (Figure 3). During systemic infections, induction of monocyte TF is a primary immune response initiated by pattern recognition receptors (PRRs) binding pathogen-associated molecular patterns (PAMPs),21-23 and/or host-derived DAMPs,24,25 or by activating immunoglobulin Fc receptors.26,27 PRR signaling supports overlapping procoagulant and proinflammatory responses in human monocytes,27 which augment TF expression.27-30 Furthermore, intracellular immune sensors such as the inflammasome promote the release of TF from pyroptotic cells,31 leading to the dissemination of procoagulant triggers. Intravascular TF expression can be further amplified by thromboinflammatory signaling through protease-activated receptors (PARs),32,33 complement mediators,34-36 P-selectin–mediated leukocyte interactions,37 and/or recognition of DAMPs.38-40 TF neutralization prevents thrombosis during experimental bacteremia41 and viremia,42,43 highlighting the critical role of TF-driven coagulation in DIC. In humans however, the essential hemostatic function of TF may preclude the systemic use of neutralizing agents, as suggested by the increased bleeding and mortality observed in a sepsis trial of active site-inhibited FVIIa.44

Initiation of coagulation in DIC. Pathogens, dead cells, and their derived molecular pattern molecules (PAMPs and DAMPs) can signal through PRRs to induce TF expression and microparticle release from monocytes, and to promote synthesis of proinflammatory cytokines that further amplify TF expression, thus initiating coagulation via the extrinsic pathway. In parallel, contact activation with FXIIa generation can occur on the surface of the pathogens, PAMPs, DAMPs, and cell debris. In particular, bacteria-derived long-chain polyphosphates (LC-poly-Ps) and platelet-derived short-chain polyphosphates (SC-poly-P), and the extracellular nucleic acids (DNA/RNA) can induce contact-mediated autoactivation of FXII and trigger coagulation via the intrinsic pathway. Both pathways converge into the common coagulation mechanism, leading to thrombin generation and downstream platelet activation and fibrin and thrombus formation.
Initiation of coagulation in DIC. Pathogens, dead cells, and their derived molecular pattern molecules (PAMPs and DAMPs) can signal through PRRs to induce TF expression and microparticle release from monocytes, and to promote synthesis of proinflammatory cytokines that further amplify TF expression, thus initiating coagulation via the extrinsic pathway. In parallel, contact activation with FXIIa generation can occur on the surface of the pathogens, PAMPs, DAMPs, and cell debris. In particular, bacteria-derived long-chain polyphosphates (LC-poly-Ps) and platelet-derived short-chain polyphosphates (SC-poly-P), and the extracellular nucleic acids (DNA/RNA) can induce contact-mediated autoactivation of FXII and trigger coagulation via the intrinsic pathway. Both pathways converge into the common coagulation mechanism, leading to thrombin generation and downstream platelet activation and fibrin and thrombus formation.
The hypercoagulant state in DIC is enhanced by dysregulation of endothelial homeostasis45 and the anticoagulant networks that control the initiation, amplification, and termination of clotting reactions. Clotting initiation is controlled by TF pathway inhibitor (TFPI), a Kunitz type inhibitor primarily exposed on endothelium and to a lesser extent on platelets and monocytes.20 In the presence of FXa, the quaternary TF-FVIIa-FXa-TFPI complex inhibits both FXa and FVIIa proteolytic activities. During the clinical course of DIC, cell-associated TFPI is cleaved by proteases,46-48 leading to a progressive increase in plasma TFPI,49 mostly truncated and devoid of anticoagulant activity. Lower active TFPI46 and accumulation of TF on “at risk” endothelial surfaces, such as branching sites, sustain procoagulant environments in septic nonhuman primates.19 Although administration of recombinant TFPI reduced DIC and mortality in experimental Gram-negative (G−) sepsis,50 TFPI supplementation in humans did not improve survival and exacerbated bleeding.51
Serpins-like C1 inhibitor and antithrombin control coagulation initiation via contact pathway by making inhibitory complexes with FXIIa and FXIa.10,12,52,53 Both serpins are consumed during DIC. Supplementation of C1 inhibitor was beneficial in preclinical sepsis models,54 whereas in humans, it reduced organ failure without improving survival.55 More recently, antibody neutralization of FXIIa or FXIa, key proteases of the contact pathway, dampened PAMP-induced coagulopathy in baboons.10,53 This therapeutic approach has yet to be tested in sepsis patients with DIC.
Amplification of coagulation
This phase is primarily regulated by antithrombin, the circulating serpin that inhibits all coagulation serine proteases.56 The anticoagulant function of antithrombin is highly enhanced by heparan-sulfate proteoglycans within the endothelial glycocalyx. Independent of its anticoagulant role, antithrombin-syndecan-4 interactions promote anti-inflammatory outcomes,57-59 reduce production of TF and proinflammatory cytokines,60 and prevent endothelial glycocalyx shedding.61 In DIC, antithrombin levels are depressed because of coagulopathic consumption, possible vascular leakage, and lower hepatic output, whereas the activity is impaired by degradation of the endothelial glycocalyx.45 Antithrombin levels below 70% associate with worsening pathology and increased mortality.56 In clinical trials, antithrombin supplementation in patients with sepsis did not improve the overall outcome, but post hoc analysis showed benefits for patients with DIC who were not concomitantly treated with heparin.62 Consequently, antithrombin supplementation was approved for sepsis-associated DIC in Japan.63
Terminal clotting reactions are downregulated by activated protein C (APC) through proteolytic inactivation of FVa and FVIIIa.64 APC anticoagulant function is enhanced by protein S and binding to anionic phospholipids where tenase and prothrombinase complexes also concentrate.65 Activation of PC requires formation of thrombin-thrombomodulin (TM) complexes on the endothelial surface and is enhanced by the endothelial PC receptor (EPCR).66 EPCR-bound APC has cytoprotective anti-inflammatory and antiapoptotic effects and prevents vascular permeability through PAR signaling.64 The PC-TM pathway is dysregulated in DIC because of shedding of TM and EPCR from endothelium67 and consumption of plasma PC.68-70 Unsurprisingly, TM and APC supplementation has been beneficial in some clinical studies. Similar to antithrombin, recombinant TM decreased mortality in patients with sepsis and DIC,71 but not in patients without DIC.63 APC has been beneficial in sepsis, despite an enhanced risk of bleeding,72 and the drug has been in clinical use for 10 years. A subsequent large clinical trial that did not show a survival benefit73 led to retraction of APC from clinical use, despite the proven advantage for patients with severe coagulopathy.74 More recently, APC variants, with decreased anticoagulant but preserved anti-inflammatory function, reduced death in murine models of sepsis and ischemic stroke64 and reduced hemorrhagic events in patients who had a stroke.75 It remains to be determined whether these APC variants can provide protection in DIC.
Inflammation-induced amplification of DIC
Pathologies with associated DIC usually exhibit systemic inflammation with pleiotropic pathophysiological effects.76 The cross talk between inflammation and hemostatic dysregulation has been extensively investigated, especially in sepsis. Proinflammatory cytokines overexpressed in DIC,77 such as tumor necrosis factor (TNF), interleukin (IL)1β, and IL6, are all procoagulant,28,30,78 whereas the regulatory IL10 attenuates clotting.79 Proinflammatory cytokines also downregulate anticoagulant mechanisms,80 support endotheliopathies,81 and modulate fibrinolysis,82,83 with a net prothrombotic effect in experimental sepsis models. In contrast to preclinical models, neither TNF84 nor IL185 inhibition reduced the incidence of DIC in sepsis trials. Similarly, IL6 blockade did not improve the coagulopathy associated with COVID-19 in a large observational cohort.86
In localized prothrombotic environments, PAR signaling can be initiated by coagulation proteases involved in the TF-dependent pathway and by APC. The outcomes of these thromboinflammatory events are complex and depend on the repertoire of PARs, the occupancy of associated receptors such as EPCRs, the differential engagement of adaptor molecules, and costimulation by inflammatory cytokines. The concept of a PAR interactome on vascular cells was proposed recently.64 In general, thrombin signaling induces proinflammatory and procoagulant responses,87 whereas APC promotes cytoprotective and anti-inflammatory outcomes.64 Of interest for DIC, thrombin induces platelet degranulation and release of procoagulant and proinflammatory proteins like FV and P-selectin,88 the latter being a potent leukocyte adhesion molecule and inducer of TF in monocytes.89 Thrombin also promotes conformational activation of integrin IIb/IIIa, which is essential for platelet aggregation,90 exposure of anionic phospholipids that enhance clotting reactions,91 and release of platelet polyphosphates,92,93 which support contact coagulation reactions. On endothelium, thrombin triggers degranulation of Weibel-Palade bodies (WPBs) and release of P-selectin and von Willebrand factor,94 induces expression of proinflammatory and cell adhesion molecules, and enhances vascular permeability.95 Inhibition of thromboinflammatory PAR1 signaling in clinical trials, albeit not DIC, reduced ischemic events but elevated the risk of bleeding.96 Biased PAR1 modulators that prevent thrombin but not APC signaling reduced thrombosis in experimental models97 and may prove beneficial in DIC as well.
Innate immune amplification of coagulopathy
DIC pathology is enhanced through immunothrombotic mechanisms that comprise primary immune responses that induce or enhance the hypercoagulant state. Excessive and/or dysregulated complement activation and neutrophil extracellular traps (NETs) amplify the coagulopathy, including that in patients with COVID-19.98 Complement is the innate proteolytic cascade tasked with recognition, extracellular killing, and clearance of invading pathogens or unprotected, damaged cells (Figure 4). Complement and coagulation pathways are closely linked through bidirectional cross talk, as detailed elsewhere.99-101 Complement mediators with direct procoagulant effects include anaphylatoxins, opsonins, and intermediates of the terminal complement complex. In general, C3a and C5a anaphylatoxins promote inflammatory activation of vascular cells, whereas C4a may alter endothelial permeability through PARs.102 C5a induces TF expression in endothelium,103 monocytes,35 and neutrophils,34 whereas terminal complement complex intermediates promote TF decryption,104,105 which is a procoagulant conformational change. Complement also activates platelets,106 leading to aggregation, exposure of anionic phospholipids, degranulation, and release of prothrombotic factors. Furthermore, C5b-9 insertion into plasma membranes promotes shedding of prothrombotic microvesicles from vascular cells,107,108 which disseminate the hypercoagulant state in DIC.109 It comes as no surprise that complement inhibitors prevent DIC development in experimental sepsis models.52,110
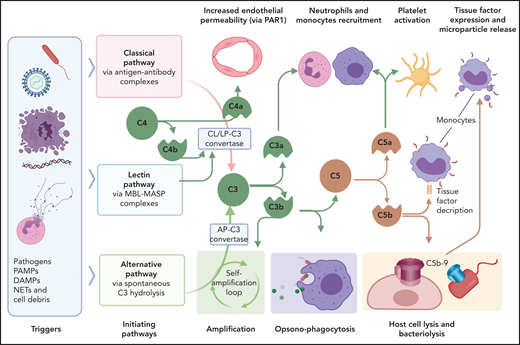
Interactions of complement with coagulation and microvasculature in DIC. Pathogens, PAMPs, NETs, cell debris, and DAMPs can activate the complement cascade via 3 pathways, classic (CP), lectin (LP), or alternative (AP), all of which converge at C3. Formation of the C3 convertase of classic and lectin pathways involves cleavage of C4 with formation of C4b convertase component and C4a, an anaphylatoxin that can increase endothelial permeability by signaling through PAR-1 and -4. C3 convertases generated through the 3 pathways, cleave C3 to C3a and C3b. C3a anaphylatoxin activates platelets and leukocytes. C3b is a potent opsonin that contributes to opsonophagocytosis of pathogens, cell debris, and circulating erythrocytes and platelets contributing to extravascular hemolysis and thrombocytopenia. C3b is also a component of C5 convertase, which cleaves C5 into C5a and C5b. C5a anaphylatoxin is a potent chemoattractant and activator of leukocytes and an inducer of TF on monocytes and PAI-1 on endothelial cells (not shown). C5b initiates the formation of C5b-9 terminal complement complex (also known as a membrane attack complex), which makes cytolytic pores in cell membranes, inducing bacteriolysis or cell death in host organs. C5b-9 can activate platelets and induce monocyte TF expression and microparticle release. MASP, mannose-associated serine protease; MBL, mannose binding lectin.
Interactions of complement with coagulation and microvasculature in DIC. Pathogens, PAMPs, NETs, cell debris, and DAMPs can activate the complement cascade via 3 pathways, classic (CP), lectin (LP), or alternative (AP), all of which converge at C3. Formation of the C3 convertase of classic and lectin pathways involves cleavage of C4 with formation of C4b convertase component and C4a, an anaphylatoxin that can increase endothelial permeability by signaling through PAR-1 and -4. C3 convertases generated through the 3 pathways, cleave C3 to C3a and C3b. C3a anaphylatoxin activates platelets and leukocytes. C3b is a potent opsonin that contributes to opsonophagocytosis of pathogens, cell debris, and circulating erythrocytes and platelets contributing to extravascular hemolysis and thrombocytopenia. C3b is also a component of C5 convertase, which cleaves C5 into C5a and C5b. C5a anaphylatoxin is a potent chemoattractant and activator of leukocytes and an inducer of TF on monocytes and PAI-1 on endothelial cells (not shown). C5b initiates the formation of C5b-9 terminal complement complex (also known as a membrane attack complex), which makes cytolytic pores in cell membranes, inducing bacteriolysis or cell death in host organs. C5b-9 can activate platelets and induce monocyte TF expression and microparticle release. MASP, mannose-associated serine protease; MBL, mannose binding lectin.
NETs are part of the innate immunity repertoire designed to trap and kill invading pathogens111 (Figure 5). This process is aided by the generation of a local fibrin mesh112 through activation of clotting reactions, dysregulation of anticoagulant networks and interaction with platelets and leukocytes.113 NETs promote clotting through TF-dependent98 and/or FXII-dependent114 pathways, whereas anticoagulant depression is mediated by neutrophil proteases concentrated on NETs.47,115 The cellular origin of TF accumulating on NETs is not always clear and may vary, depending on the underlying pathology. Exposure of P-selectin promotes NETosis,116 induces TF transcription in monocytes,37 and supports accumulation of TF+ microparticles in developing thrombi.117 Thrombin generation is enhanced by cell-free DNA released from NETs, which amplify contact pathway reactions,118 whereas histones induce platelet activation119 and WPB exocytosis.120 Not surprisingly, DNA breakdown113,121 or histone neutralization122,123 reduces the coagulopathy in experimental models. In interesting new developments, histones can substitute FVa and form an alternative prothrombinase complex with FXa,124 which escapes anticoagulant APC control. The elevated levels of circulating histones in patients with DIC124 are thus likely contributors to pathology, and their neutralization should provide therapeutic benefits.
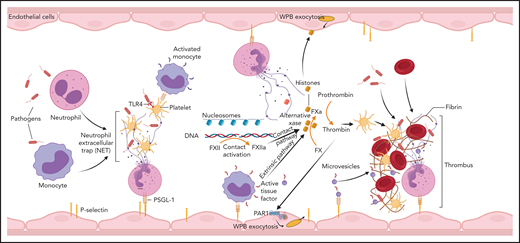
Involvement of immune mechanisms in the propagation of coagulation in DIC. Interaction of pathogens with neutrophils, macrophages, and platelets induces formation of NETs. Components of NETs, such as histones and DNA/nucleosomes, can further amplify the coagulation pathway leading to thrombin generation. NETs and proinflammatory cytokines promote TF expression on monocytes and TF-mediated coagulation. Cell-free DNA supports contact activation and intrinsic coagulation, whereas extracellular histones activate platelets and induce exocytosis of endothelial storage granules, including WPBs and subsequent release of von Willebrand factor and P-selectin, which promote microvascular thrombosis. Thrombin has a similar secretagogue effect. PSGL-1, P-selectin glycoprotein ligand-1.
Involvement of immune mechanisms in the propagation of coagulation in DIC. Interaction of pathogens with neutrophils, macrophages, and platelets induces formation of NETs. Components of NETs, such as histones and DNA/nucleosomes, can further amplify the coagulation pathway leading to thrombin generation. NETs and proinflammatory cytokines promote TF expression on monocytes and TF-mediated coagulation. Cell-free DNA supports contact activation and intrinsic coagulation, whereas extracellular histones activate platelets and induce exocytosis of endothelial storage granules, including WPBs and subsequent release of von Willebrand factor and P-selectin, which promote microvascular thrombosis. Thrombin has a similar secretagogue effect. PSGL-1, P-selectin glycoprotein ligand-1.
Fibrinolytic modulation of DIC
Intravascular fibrin initiates plasmin-mediated fibrinolysis and modulates phenotypic expression of DIC. Plasmin activation is controlled by the balanced expression of t-PA and its cognate regulator PAI-1.125 Endothelial cells are the primary source of both t-PA and PAI-1, whose synthesis is modulated by inflammatory mediators. Secretagogues such as thrombin, histamine, bradykinin, and TNF82 trigger the acute release of t-PA from endothelium, which primes fibrinolysis, whereas delayed hyperinflammatory cytokines such as IL6 induce PAI-183 expression and subsequent fibrinolytic suppression. Thrombin-stimulated platelets also release active PAI-1126 and may augment the prothrombotic state in DIC. In sepsis, elevated PAI-1 levels correlate with coagulopathy, multiple organ failure, and mortality.127 In contrast, lower PAI-1 expression128 and/or its proteolytic inactivation129 promote the hyperfibrinolytic phenotype associated with acute promyelocytic leukemia (APL). PAI-1 inhibition130 or t-PA supplementation131 attenuates thrombosis in experimental sepsis models and may be beneficial in cases of hypofibrinolysis with extremely high levels of PAI-1.132
DIC-associated pathologies
Clinical conditions with associated DIC include bacterial and viral infections and severe sterile organ damage/inflammation, such as in trauma, pancreatitis, cirrhosis, cancers, vascular abnormalities and vasculitis, heat stroke, and snake bites.2 Cumulatively, infectious diseases, trauma, and malignant disorders account for more than two-thirds of DIC cases in North America and Europe. The pathological archetype in sepsis-associated DIC is overactive clotting with suppressed fibrinolysis leading to thrombo-occlusive organ damage. Consumption of clotting factors increases the risk of bleeding in these patients. In DIC associated with trauma and obstetric calamities, severe consumption combines with abrupt hyperfibrinolysis, whereas APL and aortic aneurysms are usually associated with chronic increase of fibrinolysis and primarily lead to bleeding complications (Figure 6).

Pathogenesis of thrombotic and fibrinolytic phenotypes of DIC. DIC with a thrombotic phenotype is induced by the exposure of pathogens/PAMPs, cell debris/DAMPs, and NETs to circulating blood, leading to systemic inflammation and endothelial injury. These events promote activation of coagulation and depression of anticoagulant and fibrinolytic activities, leading to uncontrolled thrombin generation and microvascular thrombosis. When fibrinogen and other clotting factors are consumed; bleeding is a frequent outcome. Microvascular thrombosis and bleeding can be equally damaging to the tissues and organs, frequently contributing to death. DIC with fibrinolytic phenotype occurs more frequently in trauma and APL. In these cases, excessive fibrinolysis due to massive release of t-PA and plasmin generation can destroy the early hemostatic clots releasing fibrin fragment D-dimer. In APL, cell surface annexin II binds t-PA and protects its action from inhibitors, thus enhancing plasmin generation and fibrin degradation. Proteases, such as neutrophil elastase and cathepsins, released by leukocytes during inflammation and trauma, degrade fibrinogen and fibrin, leading to the formation of fibrinogen degradation products that further increase bleeding risk and contribute to organ failure and death. TAFI, thrombin activatable fibrinolysis inhibitor.
Pathogenesis of thrombotic and fibrinolytic phenotypes of DIC. DIC with a thrombotic phenotype is induced by the exposure of pathogens/PAMPs, cell debris/DAMPs, and NETs to circulating blood, leading to systemic inflammation and endothelial injury. These events promote activation of coagulation and depression of anticoagulant and fibrinolytic activities, leading to uncontrolled thrombin generation and microvascular thrombosis. When fibrinogen and other clotting factors are consumed; bleeding is a frequent outcome. Microvascular thrombosis and bleeding can be equally damaging to the tissues and organs, frequently contributing to death. DIC with fibrinolytic phenotype occurs more frequently in trauma and APL. In these cases, excessive fibrinolysis due to massive release of t-PA and plasmin generation can destroy the early hemostatic clots releasing fibrin fragment D-dimer. In APL, cell surface annexin II binds t-PA and protects its action from inhibitors, thus enhancing plasmin generation and fibrin degradation. Proteases, such as neutrophil elastase and cathepsins, released by leukocytes during inflammation and trauma, degrade fibrinogen and fibrin, leading to the formation of fibrinogen degradation products that further increase bleeding risk and contribute to organ failure and death. TAFI, thrombin activatable fibrinolysis inhibitor.
DIC in infectious diseases
Infections leading to sepsis are among the most frequent medical conditions that promote DIC. Sepsis is a life-threatening organ dysfunction syndrome caused by excessive and dysregulated host responses to microbial infection.133 Sepsis-associated DIC is a major contributor to multiple organ failure and an independent predictor of mortality.2,134
Bacterial infections
Similar DIC incidences are observed in G− and Gram-positive (G+) infections. The pathogenesis of sepsis inflammation, coagulopathy, and shock in G− bacteremia is driven by lipopolysaccharide (LPS), a specific component of the G− bacterial wall. LPS triggers TF expression in monocytes directly via CD14 - Toll-like receptor-4 (TLR4) signaling and indirectly via proinflammatory cytokines. Immunothrombotic amplification by complement,52,110 NETs,47,113,121 and DAMPs122,123 has been documented in preclinical models, but their contribution to human pathology should be confirmed in clinical studies. In G+ bacteremia, proinflammatory and procoagulant monocyte responses are triggered by TLR2 interaction with acylated cell wall PAMPs23 and/or by peptidoglycan27 activation of intracellular nucleotide-binding oligomerization domain sensors. Because of its polymeric structure, peptidoglycan also activates humoral proteolytic cascades, such as complement and the contact coagulation pathway, and impairs the anticoagulant function of antithrombin.12 The hypercoagulant state is further amplified by the release of long-chain polyphosphates from inclusion bodies of both G− and G+ bacteria. Polyphosphates can trigger the activation of FXII and downstream generation of thrombin and vasoactive bradykinin.135 Attempting to subvert immune defenses, many bacteria species secrete modulators of the host hemostasis, complement or fibrinolytic systems (Table 2), which can further influence sepsis-associated DIC.
Bacteria-derived factors that modulate coagulation, fibrinolysis, and complement systems of the host
| Bacterial factor | Bacteria species | Host target | Host effect |
|---|---|---|---|
| Staphylokinase | Staphylococcus | Plasminogen | Activation of fibrinolysis |
| Staphylocoagulase | Staphylococcus | Prothrombin | Activation of coagulation via a nonproteolytic conformational change of prothrombin |
| Clumping factors A and B (CLFA, CLFB) | Staphylococcus | Fibrinogen and platelets | Impairs pathogen clearance |
| Extracellular fibrinogen binding (Efb) protein | Staphylococcus | Fibrinogen, C3 | Inhibits platelet aggregation, neutrophil binding to fibrinogen and complement-mediated opsonization and phagocytosis |
| Staphylococcal superantigen-like protein 10 (SSL10) | Staphylococcus | Vitamin K–dependent clotting factors | Inhibits blood coagulation by targeting Gla domains of clotting factors |
| Streptokinase | Streptococcus | Plasminogen | Activation of fibrinolysis |
| Streptococcal inhibitor of complement (SIC) | Streptococcus | HK | Inhibits complement and contact phase activation |
| Surface collagen-like (Scl) proteins A and B | Group A Streptococcus | TAFI, thrombin and plasmin | Promotes TAFI activation resulting in inhibition of fibrinolysis |
| Omptins | G− bacteria (E coli, Salmonella and Yersinia) | TFPI | Inhibit TFPI-mediated anticoagulation |
| Phospholipases | Multiple bacteria species | GPI-anchored proteins | Cleaves cell surface–associated, GPI-anchored proteins (TFPI, uPAR, CD55, and CD59), thus decreasing anticoagulant, profibrinolytic, and anti-complement functions |
| Glycosidases (hyaluronidases, sialidases, heparinases, chrondroitinases) | Multiple bacteria species | Hyaluronans, sialic acid, heparin/heparan sulfate, and chondroitin sulfate residues | Degradation of the glycocalyx leading to decreased anticoagulant activity and impaired cytoprotection against histone-induced toxicity |
| DNAse | Multiple bacterial species | NETs, cell-free DNA | Degradation of prothrombotic DNA and NETs |
| Polyphosphates | Multiple bacterial species | FXII | Trigger activation of contact pathway |
| LPS | G− bacteria | CD14-TLR4 signaling; complement and contact pathway | Triggers production of proinflammatory cytokines, TF, and PAI-1 and activation of contact and alternative complement pathways |
| Peptidoglycan | G+ bacteria | FcR, NOD and inflammasome signaling; complement and contact activation pathways; inhibits antithrombin | Induces expression of cytokines and TF and the activation of complement and contact pathways |
| Bacterial factor | Bacteria species | Host target | Host effect |
|---|---|---|---|
| Staphylokinase | Staphylococcus | Plasminogen | Activation of fibrinolysis |
| Staphylocoagulase | Staphylococcus | Prothrombin | Activation of coagulation via a nonproteolytic conformational change of prothrombin |
| Clumping factors A and B (CLFA, CLFB) | Staphylococcus | Fibrinogen and platelets | Impairs pathogen clearance |
| Extracellular fibrinogen binding (Efb) protein | Staphylococcus | Fibrinogen, C3 | Inhibits platelet aggregation, neutrophil binding to fibrinogen and complement-mediated opsonization and phagocytosis |
| Staphylococcal superantigen-like protein 10 (SSL10) | Staphylococcus | Vitamin K–dependent clotting factors | Inhibits blood coagulation by targeting Gla domains of clotting factors |
| Streptokinase | Streptococcus | Plasminogen | Activation of fibrinolysis |
| Streptococcal inhibitor of complement (SIC) | Streptococcus | HK | Inhibits complement and contact phase activation |
| Surface collagen-like (Scl) proteins A and B | Group A Streptococcus | TAFI, thrombin and plasmin | Promotes TAFI activation resulting in inhibition of fibrinolysis |
| Omptins | G− bacteria (E coli, Salmonella and Yersinia) | TFPI | Inhibit TFPI-mediated anticoagulation |
| Phospholipases | Multiple bacteria species | GPI-anchored proteins | Cleaves cell surface–associated, GPI-anchored proteins (TFPI, uPAR, CD55, and CD59), thus decreasing anticoagulant, profibrinolytic, and anti-complement functions |
| Glycosidases (hyaluronidases, sialidases, heparinases, chrondroitinases) | Multiple bacteria species | Hyaluronans, sialic acid, heparin/heparan sulfate, and chondroitin sulfate residues | Degradation of the glycocalyx leading to decreased anticoagulant activity and impaired cytoprotection against histone-induced toxicity |
| DNAse | Multiple bacterial species | NETs, cell-free DNA | Degradation of prothrombotic DNA and NETs |
| Polyphosphates | Multiple bacterial species | FXII | Trigger activation of contact pathway |
| LPS | G− bacteria | CD14-TLR4 signaling; complement and contact pathway | Triggers production of proinflammatory cytokines, TF, and PAI-1 and activation of contact and alternative complement pathways |
| Peptidoglycan | G+ bacteria | FcR, NOD and inflammasome signaling; complement and contact activation pathways; inhibits antithrombin | Induces expression of cytokines and TF and the activation of complement and contact pathways |
Viral infections
Coagulopathic complications commonly develop during acute infections with Ebola, Marburg, Crimean-Congo, Lassa, yellow fever, hantavirus, dengue,136 and, more recently, the SARS-CoV-2 viruses.137 Virus PAMPs activate endosomal TLRs, including TLR3 and TLR7, which also promote TF transcription.22,138 Viruses trigger TF expression in endothelium139,140 and monocytes,141-143 and TF neutralization reduces coagulopathy and improves survival in Ebola42 and HIV43 infections. Similar to bacteremia, TF+ monocytes also express the proinflammatory cytokines TNF, IL1β, and IL6.43 These procoagulant and proinflammatory monocytes persist after virological suppression43 and may support coagulopathic complications after treatment of the underlying disease. Enveloped viruses such as herpes simplex can also capture host-derived TF in their phospholipid envelope. The TF+ virions exhibit enhanced infectivity,144 whereas the exposed TF is procoagulant145 and may disseminate the hypercoagulant state. Overall, viremia-associated DIC usually exhibits a thrombotic phenotype that can lead to hemorrhage due to consumption of clotting factors, thrombocytopenia, rapid decrease of TM-PC anticoagulants, and dysregulated fibrinolysis.
The intravascular coagulopathy commonly observed in severe SARS-CoV-2 infections, which frequently exhibit nonovert DIC, can trigger both thrombotic and bleeding complications.137 The progressive D-dimer elevation documented in nonsurviving patients with COVID-19146 indicates active coagulopathy, associates with hyperinflammatory markers, and predicts thrombotic complications and mortality.137,146 Elevated D-dimer accompanied by thrombocytopenia predict an enhanced bleeding risk.137 No significant changes in global coagulation test results and fibrinogen level were observed between patients with or without coagulopathic COVID-19,137 indicating compensatory hepatic synthesis of clotting factors. The coagulopathic mechanisms in COVID-19 are under active investigation. Because endothelial cells harbor the ACE-2 receptor,147 SARS-CoV-2 can directly infect the endothelium promoting endotheliopathy and microvascular thrombosis.148,149 Unlike SARS-CoV-1, which replicates and triggers TF transcription in peripheral blood mononuclear cells,143 SARS-CoV-2 replication in monocytes has not been confirmed. Nevertheless, intravascular TF can be induced by platelet-monocyte aggregates in patients with COVID-19.150 We learned that inflammatory mediators,151 complement,152 and autoimmune mechanisms153 could amplify the coagulopathy, but such findings will require mechanistic confirmation in experimental models. Thus, anticoagulants can benefit patients with prothrombotic COVID-19,154 but coagulopathic monitoring is still necessary to prevent bleeding.137 At the moment, the American Society of Hematology recommends prophylactic-intensity anticoagulants for patients with COVID-19 who do not have confirmed venous thromboembolism.155
DIC in noninfectious diseases
Coagulopathies associated with most cancers are TF dependent.156,157 TF expression by malignant cells and cancer-derived microparticles contributes to tumor progression and metastasis.158 Some tumors also express a cysteine endopeptidase, cancer procoagulant, which activates FX directly and impairs hemostasis.159 In addition, circulating carcinoma mucins activate platelets and support prothrombotic, selectin-mediated platelet-leukocyte interactions.160,161 Cancer-associated coagulopathy usually exhibits a chronic asymptomatic progression, where clotting factors and platelets are compensated for. Thromboembolic manifestations are more commonly seen with solid tumors, whereas hemorrhagic events predominate in hematologic malignancies.157 Consumptive coagulopathy or dysregulated activation of plasminogen by annexin II162 exacerbate bleeding in APL. Whereas patients with metastatic malignancies that display slow-onset thrombotic DIC can benefit from prophylactic heparins, antifibrinolytic agents have been used to limit hemorrhages in hyperfibrinolytic APL patients.157
The coagulopathy of trauma is triggered by the exposure of perivascular TF after injury and is amplified by DAMPs, hemorrhagic shock, and potential superimposed infection resulting in thrombin generation, platelet activation, and fibrinolytic dysfunction.163 In early trauma, DIC manifests a hyperfibrinolytic phenotype due to excessive t-PA release from endothelial cells. Consequently, antifibrinolytic agents, such as tranexamic acid, were beneficial when administered during the first 3 hours.164 At later stages, the hypofibrinolytic shift caused by delayed PAI-1 expression promotes microvascular thrombosis and contributes to organ damage. Delayed administration of tranexamic acid exacerbated the thrombo-occlusive phenotype and increased the risk of death.164
DIC complications are also observed in pregnancy and obstetric emergencies.165 Abruptio placentae, amniotic fluid embolism, and eclampsia lead to leakage of TF from the placenta into the maternal circulation during labor, which, coupled with reduced expression of TM166 and elevation of PAI-1, can increase the risk of thrombotic complications.167 Neonates are also predisposed to coagulopathies related to their incompletely developed hemostatic system. As a result, DIC incidence is higher in premature babies with underlying conditions such as sepsis, acute respiratory distress syndrome, intravascular hemolysis, and necrotizing enterocolitis.168
Future perspectives
The diversity of DIC, in terms of underlying conditions, prothrombotic triggers, anticoagulant dysregulation, and the gamut of hemostatic complications from thrombosis to bleeding that can sometimes occur in the same patient during the course of disease, makes it challenging to assess this broad pathology in clinical settings. As a result, nonovert DIC states are harder to diagnose accurately, and overt DIC diagnosis is usually confirmed when physiologic compensatory mechanisms are long overcome. Better biomarker definitions and stratification of DIC subtypes and patients are needed. Ideally, combinations of biomarkers that define DIC subtypes would reflect the underlying pathologic mechanisms and allow for targeted therapeutic approaches, unlike the global anticoagulant strategies used so far. This need is painfully obvious from retrospective analyses of anticoagulant trials in sepsis, where benefits observed ex post facto in subgroups of patients are drowned by the overall heterogeneity of these studies. Furthermore, we need a better understanding of the pre-DIC state along with predictive assessments of clinical developments. This knowledge would greatly expand the therapeutic repertoire, adding preventive interventions with fewer side effects.
Current therapeutic approaches in patients with DIC target the resolution of the underlying trigger (infection, trauma, and malignancy) and are mostly compensatory in nature, being restricted to supplementation of the missing components (clotting factors, anticoagulants, and platelets) necessary to restore hemostatic balance.
Targeting the TF pathway directly or by TFPI supplementation significantly increases the risk of bleeding due to hemostatic impairment. Unless the inhibitors can be spatially and/or temporally aimed toward the developing thrombi, they are unlikely to be useful systemically. In contrast, limiting clotting amplification through inhibition of the contact/intrinsic pathways and/or modulation of terminal reactions seems to have better potential. We and others have shown that contact pathway inhibition reduces sepsis-associated coagulopathy and improves organ function in experimental bacteremia.10,53 Defining the role of the contact pathway in DIC associated with other pathologies is needed to identify additional patients for whom the strategy may be useful. Likewise, inhibitors of terminal clotting reactions have long been used for thromboprophylaxis. They usually have relatively long half-lives in circulation, and, as such, their use is problematic in fast-progressing acute coagulopathies with the potential for bleeding. In contrast, short-lived inhibitors of FXa and thrombin attenuated DIC and protected organ function in Escherichia coli sepsis in baboons,169 and may be useful in clinical settings. These strategies nevertheless require further clinical validation.
Last, the role of genomic variation in the incidence and phenotypic expression of DIC has been underinvestigated to date. With the expansion of genomic sequencing, we expect associations between polymorphisms and DIC to multiply during the next decade, at least in cancer-associated DIC. Integration of genetic data, biomarker dynamics, and clinical outcomes will not be trivial and is likely to require machine learning algorithms and collaboration of computer and basic science and clinical researchers to tailor individualized therapies for patients.
Acknowledgments
The authors thank Fletcher B. Taylor and Gary Kinasewitz for critical reading of the manuscript. The authors apologize to colleagues whose work could not be cited because of space constraints. The Illustrations were created with BioRender.com.
Investigations of coagulopathies by our group were supported by grants from the National Institutes of Health, National Institute of General Medical Sciences (GM116184, GM12160, and GM122725) (F.L.), and by National Institute of Allergy and Infectious Diseases grants AI157037 (F.L.) and U19AI062629 (F.L. and N.I.P.).
Authorship
Contribution: All authors drafted the first version of different sections of the manuscript and critically reviewed the final manuscript.
Conflict-of-interest disclosure: F.L. has received research contract funding from Bayer and Ra Pharma. The remaining authors declare no competing financial interests.
Correspondence: Florea Lupu, Cardiovascular Biology Research Program, Oklahoma Medical Research Foundation, 825 NE13th St, Oklahoma City, OK 73104; e-mail: florea-lupu@omrf.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal