

Key Points
GALEN regimen is a potent regimen in 1st FL with 94% ORR and 80% CCR according to the 2014 Lugano post hoc analysis and 82% 3-year PFS.
Toxicity is concordant with previous studies in relapsed or refractory setting and comprises mainly neutropenia (47% grade ≥3).

Abstract
Obinutuzumab and lenalidomide (referred to as the GALEN combination) is an active immunomodulatory combination with a manageable safety profile in multiple types of lymphoma. We report efficacy and safety results for the phase 2 GALEN study in previously untreated patients with advanced follicular lymphoma (FL). Eligible patients aged ≥18 years had an Eastern Cooperative Oncology Group performance status ≤2 and high-tumor burden, grade 1 to 3a FL. Induction treatment was obinutuzumab (1000 mg IV, days 8, 15, and 22, cycle 1; day 1, cycles 2-6) plus lenalidomide (20 mg/d, days 1-21, cycle 1; days 2-22, cycles 2-6) for six 28-day cycles. Maintenance included obinutuzumab (1000 mg every 2 cycles) plus lenalidomide (10 mg, days 2-22) for ≤12 cycles (year 1) followed by obinutuzumab (1000 mg every 56 days) for 6 cycles (year 2). The primary end point was complete response rate (CRR) after induction per the 1999 International Working Group criteria. From October 2015 to February 2017, a total of 100 patients were enrolled. CRR after induction was 47%, and the overall response rate (ORR) was 92%. Post hoc analyses per the 2014 Lugano classification, including patients with missing bone marrow assessments, identified an additional 13 patients fulfilling CRR criteria, resulting in a complete metabolic response of 80% and an ORR of 94%. At a median follow-up of 3.7 years, 3-year progression-free survival and overall survival were 82% and 94%, respectively. The most common adverse event was neutropenia (48% any grade; 47% grade ≥3). Only 2% of patients presented with febrile neutropenia; others were mainly grade ≤2. No other specific grade ≥3 toxicity occurred at a frequency >3%. Overall, these results showed promising clinical efficacy for the chemotherapy-free GALEN backbone in previously untreated patients with high tumor burden FL. Except for neutropenia, the safety profile of the combination is remarkable. The study was registered at clinicaltrials.gov as #NCT01582776.
Introduction
Follicular lymphoma (FL) is the most common indolent lymphoma, accounting for ∼30% of cases.1 Over the 2 last decades, the addition of rituximab in combination with chemotherapy has significantly prolonged overall survival (OS), and maintenance therapy led to an almost doubled progression-free survival (PFS) after first-line induction.2,3
Lenalidomide has exhibited multiple immunomodulatory properties involving, among others, cytokine production alteration, amplified T-cell costimulation, and increased natural killer cell cytotoxicity.4-6 In vitro experiments reported improved antibody-dependent, cell-mediated cytotoxicity and potent synergy between rituximab and lenalidomide. In patients with FL, the combination of lenalidomide and rituximab proved to be effective in the relapsed or refractory (R/R) setting and was recently approved given its significantly prolonged PFS over rituximab monotherapy.7-9 As first-line therapy, lenalidomide and rituximab have produced efficacy results that seem comparable to those of immunochemotherapy, with less toxicity.10-12
Obinutuzumab is an immunoglobulin G1–type recombinant, humanized, and glycoengineered type II anti-CD20 antibody.13 Compared with rituximab, obinutuzumab mediates greater antibody-dependent cellular cytotoxicity with reduced complement-dependent cytotoxicity.13,14 In FL, obinutuzumab in combination with chemotherapy showed significantly prolonged PFS compared with first-line rituximab-based treatment and is now used worldwide.15
Given these observations, our cooperative group hypothesized that obinutuzumab and lenalidomide (thereafter referred to as the GALEN combination) would represent an attractive first-line treatment strategy for patients with FL to improve immune effector cell mechanisms. The recommended phase 2 dose of 20 mg for lenalidomide, when associated with the 1000-mg fixed-dose obinutuzumab, was previously defined by our group in a phase 1b study.16 In patients with R/R FL, this combination was safe and effective, with 68 (79%) of 86 evaluable patients responding at the end of the 6-month induction.17 At 2 years, PFS, duration of response (DOR), and OS were 65%, 70%, and 87%, respectively. No unexpected safety signals emerged, and neutropenia was the most frequent grade ≥3 toxicity, with only 5% of patients experiencing febrile neutropenia.
Here, we report the phase 2 study results assessing the efficacy and safety of the GALEN combination in patients with previously untreated FL.
Patients and methods
Patients
In this international multicenter phase 2 study, eligible patients were ≥18 years of age with biopsy-proven grade 1, 2, or 3a, CD20-positive FL, Eastern Cooperative Oncology Group performance status ≤2, and no prior systemic treatment of lymphoma. Patients had to have a high tumor burden according to GELF (Groupe d’Etude des Lymphomes Folliculaires) criteria as defined by at least one of the following criteria: one lymphoma lesion >7 cm, ≥3 separate nodes of ≥3 cm, symptomatic splenic enlargement or organ compression by lymphoma, the presence of B symptoms, pleural or peritoneal effusion, or lactate dehydrogenase or β2-microglobulin levels above the upper limit of normal. Patients were required to comply with pregnancy prevention requirements, present with life expectancy ≥3 months, and have measurable disease defined by ≥1 measurable lesion on computed tomography (CT) scan (transverse diameter >1.5 cm and short-axis ≥10 mm). Exclusion criteria were the presence of central nervous system involvement, known HIV or human T-cell leukemia virus type 1, or active hepatitis B or C viral infection. Patients with any serious active disease or comorbid medical condition or pulmonary disease, or with a history of malignancies other than lymphoma unless cancer-free for ≥5 years, were ineligible. The use of corticosteroids ≥10 mg/d within the previous 28 days was prohibited. A prior watch-and-wait period was not considered as an exclusion criterion.
Histologic diagnosis of FL was made by a local pathologist before inclusion. A centralized pathology review was performed by a pathology panel comprising ≥2 expert hematopathologists.
The study protocol was approved by local or national ethics committees according to the laws of each country and undertaken in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. Patients were required to provide written informed consent before registration.
Treatment
All patients received an induction phase of six 28-day cycles with a combination of obinutuzumab (8 intravenous infusions of a 1000-mg flat dose on days 8, 15, and 22 of cycle 1 and on day 1 for cycles 2-6) and lenalidomide (20 mg/d on days 1-21 of cycle 1 and days 2-22 for cycles 2-6) as previously described.17 Subsequently, patients who achieved at least a partial response (PR) according to the 1999 International Working Group (IWG) criteria18 after 6 cycles received maintenance treatment. During the first year of maintenance (12 cycles of 28 days), patients received obinutuzumab (6 total infusions of 1000 mg, 1 per every 2 cycles of 28 days each) and lenalidomide (10 mg on days 2-22 of each 28-day for ≤12 cycles) until disease progression or treatment discontinuation. During the second year of maintenance (6 cycles of 56 days), patients received obinutuzumab (6 infusions of 1000 mg every 56 days) until disease progression or treatment discontinuation. The treatment schedule is summarized in supplemental Figure 1 (available on the Blood Web site).
All patients were administered a daily aspirin (100 mg) for deep vein thrombosis prophylaxis during treatment through 28 days after the end of lenalidomide treatment. Patients unable to tolerate aspirin and those with a history of deep vein thrombosis or deemed high risk received low-molecular-weight heparin or warfarin (Coumadin, Bristol Myers Squibb). Granulocyte colony-stimulating factor was permitted according to American Society of Clinical Oncology and European Society of Medical Oncology guidelines and recommended if the absolute neutrophil count was <500/mm3 within any cycle. Treatment with granulocyte colony-stimulating factor up to 3 days was allowed to reach the absolute neutrophil count required to administer the next cycle.
The percentage of planned dose was calculated as follows: (total dose taken in milligrams)/(total dose expected in milligrams) × 100. It was computed for each drug and each treatment period (induction and maintenance).
Response assessment
CT imaging of the chest, abdomen, and pelvis with intravenous contrast was performed at baseline, after 3 cycles, at the end of induction, every 6 months during the 2 years of maintenance therapy, and thereafter every 6 months during follow-up, or until disease progression or relapse. Positron emission tomography (PET)/CT imaging was optional but performed for all patients at baseline and virtually all patients at the end of induction. Only PET imaging for patients with a Deauville score ≥3 (n = 32) or with a missing Deauville score (n = 4) according to local evaluation at the end of induction were centrally reviewed. Patients with a local Deauville score ≤2 (n = 61) were considered in complete metabolic response (CMR) without the need for central review. Patients with no PET imaging at the end of induction (n = 3) were not evaluable.
Statistical analysis
The primary end point was the complete response rate (CRR; complete response [CR] plus unconfirmed CR [CRu]) according to the 1999 IWG criteria18 at the end of 6 cycles of GALEN. Secondary end points for response according to IWG 1999 criteria were overall response rate (ORR) at the end of induction, ORR and CRR at the end of treatment (induction plus maintenance), and best response during treatment. PFS, time to next antilymphoma treatment (TNALT), DOR, OS, and safety were also secondary end points. Response rates according to the Lugano 2014 classification19 at the end of induction were exploratory, post hoc analyses not initially planned since the trial design was initiated in 2012. Progression of disease during the first 24 months after treatment start (ie, POD24) was also a post hoc analysis.
The trial was designed to detect a CRR increase from 50% (null hypothesis) to 65% (alternative hypothesis) assuming a 90% power at an overall 5% (one-sided) significance level using a single-stage phase 2 design. Based on these hypotheses, the null hypothesis (CRR ≤50%) would be rejected if the lower limit of the 90% confidence interval (CI) was ≥50%, meaning that at least 59 patients should be in CR at the end of induction. Considering a 7% dropout rate, 100 patients needed to be included according to sample size computation. Adverse events (AEs) were evaluated in accordance with the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03. Analyses were conducted by using SAS version 9.3 (SAS Institute, Inc.). The study was registered with clinicaltrials.gov as #NCT01582776.
Results
Patients
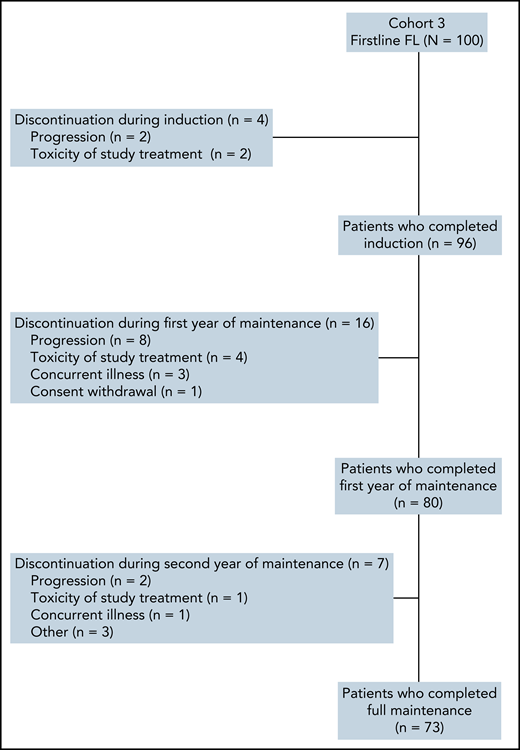
From October 2015 to February 2017, a total of 100 patients were enrolled in 21 centers from France and Belgium (Figure 1). All patients were evaluable for efficacy and safety analyses. Ninety-six (96%) patients completed all 6 cycles of induction. Four patients discontinued treatment during induction due to progression (n = 2) or toxicity (n = 2). Seventy-three patients (73%) completed 2 years of maintenance treatment. Sixteen patients discontinued during the first year of obinutuzumab and lenalidomide maintenance (8 for progression, 4 for toxicity, and 4 for other reasons) and 7 during the second year of obinutuzumab-only maintenance (2 for progression, 1 for toxicity and 4 for other reasons).
Patient characteristics are summarized in Table 1. Briefly, median age at enrollment was 60.5 years (range, 32-89 years), 45% were male, and 75% had Eastern Cooperative Oncology Group performance status 0. With regard to risk factors, 17%, 40%, and 43% of patients had a low, intermediate, or high Follicular Lymphoma International Prognostic Index score, respectively. Most patients had FL grade 1 to 2 (91%). Of note, nearly one-third of patients presented with bulky FL (ie, largest diameter >7 cm).
Baseline patient characteristics (N = 100)
| Characteristic | Value |
|---|---|
| Age at inclusion, median (range), y | 60.5 (32-89) |
| Age group | |
| ≤70 y | 83 (83) |
| >70 y | 17 (17) |
| Sex | |
| Male | 45 (45) |
| Female | 55 (55) |
| ECOG PS | |
| 0 | 75 (75) |
| 1 | 24 (24) |
| 2 | 1 (1) |
| Ann Arbor stage | |
| II | 11 (11) |
| III | 22 (22) |
| IV | 67 (67) |
| Highest diameter >7 cm | |
| No | 69 (69) |
| Yes | 31 (31) |
| Time between biopsy of initial diagnosis and inclusion, median (IQR), mo | 1.6 (1-3) |
| Initial histologic diagnosis (per local assessment) | |
| CD20+ FL grade 2 | 72 (72) |
| CD20+ FL grade 1 | 13 (13) |
| CD20+ FL grade 3a | 9 (9) |
| Other | 6 (6) |
| CD20+ FL grades 1-2 | 5 (5) |
| Low-grade FL | 1 (1) |
| FLIPI group | |
| Low | 17 (17) |
| Intermediate | 40 (40) |
| High | 43 (43) |
| LDH levels >ULN | |
| No | 73 (73) |
| Yes | 27 (27) |
| B symptoms | |
| No | 82 (82) |
| Yes | 18 (18) |
| BM involvement* | |
| No | 47 (52) |
| Yes | 43 (48) |
| Missing | 10 |
| β2-microglobulin levels >ULN* | |
| No | 50 (52) |
| Yes | 46 (48) |
| Missing | 4 |
| Characteristic | Value |
|---|---|
| Age at inclusion, median (range), y | 60.5 (32-89) |
| Age group | |
| ≤70 y | 83 (83) |
| >70 y | 17 (17) |
| Sex | |
| Male | 45 (45) |
| Female | 55 (55) |
| ECOG PS | |
| 0 | 75 (75) |
| 1 | 24 (24) |
| 2 | 1 (1) |
| Ann Arbor stage | |
| II | 11 (11) |
| III | 22 (22) |
| IV | 67 (67) |
| Highest diameter >7 cm | |
| No | 69 (69) |
| Yes | 31 (31) |
| Time between biopsy of initial diagnosis and inclusion, median (IQR), mo | 1.6 (1-3) |
| Initial histologic diagnosis (per local assessment) | |
| CD20+ FL grade 2 | 72 (72) |
| CD20+ FL grade 1 | 13 (13) |
| CD20+ FL grade 3a | 9 (9) |
| Other | 6 (6) |
| CD20+ FL grades 1-2 | 5 (5) |
| Low-grade FL | 1 (1) |
| FLIPI group | |
| Low | 17 (17) |
| Intermediate | 40 (40) |
| High | 43 (43) |
| LDH levels >ULN | |
| No | 73 (73) |
| Yes | 27 (27) |
| B symptoms | |
| No | 82 (82) |
| Yes | 18 (18) |
| BM involvement* | |
| No | 47 (52) |
| Yes | 43 (48) |
| Missing | 10 |
| β2-microglobulin levels >ULN* | |
| No | 50 (52) |
| Yes | 46 (48) |
| Missing | 4 |
Data are presented as no. (%) unless otherwise specified. ECOG PS, Eastern Cooperative Oncology Group performance status; FLIPI, Follicular Lymphoma International Prognostic Index; IQR, interquartile range; LDH, lactate dehydrogenase; ULN, upper limit of normal.
Calculated percentages excluded missing data.
Seventy-nine percent and 89% of patients received >90% of the planned lenalidomide and obinutuzumab doses, respectively, during the six 28-day cycles of the induction phase. Including the maintenance phase, the percentage of planned doses for induction followed by maintenance was >90% for 56% and 76% of patients receiving lenalidomide and obinutuzumab.
Efficacy
The CRR at the end of induction according to the 1999 IWG criteria18 (primary end point) was 47% (90% CI [primary end point evaluation], 38-56; 95% CI, 37-57), with 20% CR and 27% CRu (Table 2). The PR rate was 45%, leading to an ORR of 92% (95% CI, 85-96). Secondary end points of CRR and ORR at the end of treatment (induction plus maintenance, ie CR after 30 months [CR30]) were 63% (95% CI, 52-72) and 79% (95% CI, 70-87), respectively. Best CRR and ORR during the entire treatment duration were 77% (95% CI, 68-85) and 99% (95% CI, 95-100).
Response rates according to the 1999 IWG criteria18 at the end of induction (primary end point), end of induction and maintenance, and best response rates
| Response status* | End of induction (primary end point) (N = 100) | End of induction and maintenance† (n = 96) | Best response rates (N = 100) |
|---|---|---|---|
| ORR‡ | 92 (92) [85-96] | 76 (79) [70-87] | 99 (99) [95-100] |
| CRR§ | 47 (47) [37-57] | 60 (63) [52-72] | 77 (77) [68-85] |
| CR | 20 (20) | 44 (46) | 51 (51) |
| CRu | 27 (27) | 16 (17) | 26 (26) |
| PR | 45 (45) | 16 (17) | 22 (22) |
| Stable disease | 1 (1) | 0 (0) | 0 (0) |
| Progressive disease | 2 (2) | 9 (9) | 1 (1) |
| Not evaluated | 5 (5) | 11 (11) | 0 (0) |
| Response status* | End of induction (primary end point) (N = 100) | End of induction and maintenance† (n = 96) | Best response rates (N = 100) |
|---|---|---|---|
| ORR‡ | 92 (92) [85-96] | 76 (79) [70-87] | 99 (99) [95-100] |
| CRR§ | 47 (47) [37-57] | 60 (63) [52-72] | 77 (77) [68-85] |
| CR | 20 (20) | 44 (46) | 51 (51) |
| CRu | 27 (27) | 16 (17) | 26 (26) |
| PR | 45 (45) | 16 (17) | 22 (22) |
| Stable disease | 1 (1) | 0 (0) | 0 (0) |
| Progressive disease | 2 (2) | 9 (9) | 1 (1) |
| Not evaluated | 5 (5) | 11 (11) | 0 (0) |
Data are presented as no. (%) [95% CI] or no. (%).
Patients without response assessment (due to whatever reason) were considered nonresponders.
Period of evaluation was after complete treatment of 74 patients (77%) and at premature withdrawal for 22 patients (23%).
ORR = (CR + CRu + PR).
CRR = (CR + Cru).
Thirty-two patients with no bone marrow (BM) biopsy or with marrow involvement at baseline did not have a BM biopsy performed at the end of induction. Among them, 17 patients fulfilling CR or CRu criteria, based on reduction of tumor size by CT scan per 1999 IWG criteria,18 had to be reclassified as PR due to missing BM evaluation at the end of induction (supplemental Table 1). According to the 2014 Lugano classification,19 13 of these 17 patients were in CMR at the end of induction. As a result, CR according to the 2014 Lugano classification at the end of induction was 80% (95% CI, 71-87) and overall response was 94% (95% CI, 87-98) (Table 3). Of note, CR at the end of treatment (ie, after 30 months [CR30]) was 63% (95% CI, 52-62) according to Cheson 99 criteria.
Response rates at the end of induction according to the 2014 Lugano response classification (exploratory analysis) (N = 100)
| Response status* | Value |
|---|---|
| Overall metabolic response | 94 (94) [87-98] |
| Complete metabolic response | 80 (80) [71-87] |
| Partial metabolic response | 14 (14) |
| No metabolic response (or stable disease) | 2 (2) |
| Progressive metabolic disease | 0 (0) |
| Not evaluable | 4 (4) |
| Response status* | Value |
|---|---|
| Overall metabolic response | 94 (94) [87-98] |
| Complete metabolic response | 80 (80) [71-87] |
| Partial metabolic response | 14 (14) |
| No metabolic response (or stable disease) | 2 (2) |
| Progressive metabolic disease | 0 (0) |
| Not evaluable | 4 (4) |
Data are presented as no. (%) [95% CI] or no. (%).
Defined based on a 5-point scale according to PET/CT-guided assessments as outlined by the 2014 Lugano revised criteria for response classifications.19
After a median follow-up of 3.7 years (95% CI, 3.6-3.8), estimated 3-year PFS was 82% (95% CI, 73-88), TNALT was 85% (95% CI, 76-91), DOR was 83% (95% CI, 74-89), and OS was 94% (95% CI, 87-97) (Figure 2).
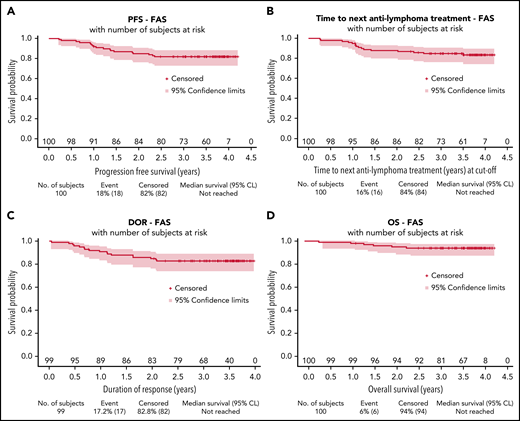
Survival distributions for the full analysis set. PFS (A), TNALT (B), response duration (C), and OS (D). CL, confidence limit; FAS, full analysis set.
Survival distributions for the full analysis set. PFS (A), TNALT (B), response duration (C), and OS (D). CL, confidence limit; FAS, full analysis set.
No significant differences in response rate or PFS duration were observed according to lenalidomide dose intensity (<90% or ≥90%) received during induction (supplemental Table 2; supplemental Figure 2). PFS was also consistent across all patient subgroups (supplemental Figure 3).
Reaching a CMR according to the 2014 Lugano criteria was associated with significantly prolonged PFS and TNALT (P < .001 for both) (supplemental Figure 4A-B). CR/CRu status, however, was not predictive of a better outcome, most likely due to the high proportion of patients considered in PR according to the Cheson 99 criteria (because of lack of BM reassessment) despite reaching CMR according to PET-CT evaluation (supplemental Figure 4C-D).
Of 98 patients evaluable for early progression (1 patient died of unrelated cancer without progression, and 1 patient withdrew consent before 2 years), 14 patients (14%) experienced early POD24 after treatment initiation. Biopsy was performed at progression in 10 cases, and histologic transformation occurred in 3 patients. Patients experiencing POD24 were confirmed to have a shorter subsequent OS (P < .0001) (supplemental Figure 5).
Safety
The most frequently reported AEs of any grade during treatment were asthenia (51%), neutropenia (48%), constipation (42%), cough (36%), and diarrhea (35%) (Table 4). The vast majority of these AEs were grade ≤2, except for neutropenia. Forty-seven percent of patients experienced grade ≥3 neutropenia, but only 2 patients presented with febrile neutropenia. The second most frequent grade ≥3 AE was infusion-related reaction, which occurred in only 3% of patients. Twenty-one patients experienced at least 1 serious AE during induction, 30 patients during maintenance, and 14 patients after treatment. Treatment discontinuation occurred in 75 patients. The most frequent reason for treatment discontinuation was neutropenia (37% of patients) and infection (20%), followed by infusion-related reaction (17%) and skin reaction (14%). Tumor flare was observed in only 4% of patients, and only 1% was of grade 3. Seven patients permanently discontinued treatment due to toxicity (Figure 1). Thirty-two patients had lenalidomide dose modification, and the main reason was neutropenia (26% of patients).
AEs (safety population [N = 100])
| AE | Any grade | Grade ≥3 |
|---|---|---|
| All AEs | 100 (100) | 74 (74) |
| Asthenia | 51 (51) | 2 (2) |
| Neutropenia* | 48 (48) | 47 (47) |
| Constipation | 42 (42) | 0 (0) |
| Cough | 36 (36) | 2 (2) |
| Diarrhea | 35 (35) | 2 (2) |
| Infusion-related reaction | 28 (28) | 3 (3) |
| Bronchitis | 27 (27) | 0 (0) |
| Decreased weight | 26 (26) | 1 (1) |
| Arthralgia | 25 (25) | 0 (0) |
| Rash | 25 (25) | 2 (2) |
| Rhinitis | 21 (21) | 0 (0) |
| Nausea | 18 (18) | 1 (1) |
| Nasopharyngitis | 17 (17) | 0 (0) |
| Back pain | 15 (15) | 0 (0) |
| Pruritus | 15 (15) | 1 (1) |
| Urinary tract infection | 15 (15) | 0 (0) |
| Peripheral edema | 13 (13) | 1 (1) |
| Muscle spasms | 12 (12) | 1 (1) |
| Upper abdominal pain | 11 (11) | 0 (0) |
| Erythema | 11 (11) | 0 (0) |
| Peripheral neuropathy† | 11 (11) | 1 (1) |
| Sinusitis | 11 (11) | 0 (0) |
| AE | Any grade | Grade ≥3 |
|---|---|---|
| All AEs | 100 (100) | 74 (74) |
| Asthenia | 51 (51) | 2 (2) |
| Neutropenia* | 48 (48) | 47 (47) |
| Constipation | 42 (42) | 0 (0) |
| Cough | 36 (36) | 2 (2) |
| Diarrhea | 35 (35) | 2 (2) |
| Infusion-related reaction | 28 (28) | 3 (3) |
| Bronchitis | 27 (27) | 0 (0) |
| Decreased weight | 26 (26) | 1 (1) |
| Arthralgia | 25 (25) | 0 (0) |
| Rash | 25 (25) | 2 (2) |
| Rhinitis | 21 (21) | 0 (0) |
| Nausea | 18 (18) | 1 (1) |
| Nasopharyngitis | 17 (17) | 0 (0) |
| Back pain | 15 (15) | 0 (0) |
| Pruritus | 15 (15) | 1 (1) |
| Urinary tract infection | 15 (15) | 0 (0) |
| Peripheral edema | 13 (13) | 1 (1) |
| Muscle spasms | 12 (12) | 1 (1) |
| Upper abdominal pain | 11 (11) | 0 (0) |
| Erythema | 11 (11) | 0 (0) |
| Peripheral neuropathy† | 11 (11) | 1 (1) |
| Sinusitis | 11 (11) | 0 (0) |
Data are presented as no. (%).
Included combined preferred terms: neutropenia, neutrophil count decreased, neutrophil percentage decreased, and band neutrophil count decreased.
Included combined preferred terms: neuropathy peripheral, peripheral sensory neuropathy, and paresthesia.
Regarding AEs of infectious nature, 75% of patients experienced at least one infection throughout treatment, and most of them concerned the respiratory tract. No specific period of occurrence (ie, induction, first or second year of maintenance therapy) was observed. When all AEs of infectious nature were grouped, only 8% of patients experienced grade ≥3 infectious AEs.
Three fatal AEs were observed. A 78-year-old woman died of an intestinal adenocarcinoma during the first year of maintenance, a 68-year-old man died of a lung adenocarcinoma after treatment completion, and a 70-year-old man died of septic shock after cycle 4 of induction. All of these deaths were considered unrelated to lenalidomide or obinutuzumab by the local investigators.
Eleven patients presented with a second primary malignancy (SPM) (supplemental Table 3; supplemental Figure 6). One patient presented with 2 sequential second primary malignancies (lung adenocarcinoma followed by breast cancer). Altogether, 10 solid tumors and 2 hematologic malignancies were noted. Two of these were considered unrelated to study treatment by local investigators. One patient had a lung adenocarcinoma diagnosed 6 months after inclusion, but pulmonary nodules were present on baseline CT scan. One patient presented with a neuroendocrine pancreatic tumor diagnosed 7 months after inclusion, but the pancreatic tumor mass was already present at baseline. The 2 hematologic SPMs were a Hodgkin lymphoma diagnosed 13 months after inclusion in a patient achieving a CRu after GALEN induction, and a peripheral T-cell lymphoma not otherwise specified, also diagnosed 13 months after inclusion in a patient achieving CR after induction. Finally, 2 SPMs were cutaneous basocellular and epidermoid carcinomas. A detailed list of other SPMs is provided in supplemental Table 3.
Discussion
Results of the study showed that obinutuzumab and lenalidomide in combination for 6 months, followed by a first year of maintenance with both drugs and a second year of maintenance with obinutuzumab only, is active and safe in patients with previously untreated, high tumor burden FL. The primary end point of this phase 2 study (ie, CRR at the end of six 28-day cycles of GALEN) was not reached likely due to the missing BM assessments at the end of induction to confirm CR or CRu in 17 patients, even though the procedure was required per protocol, thus leading to their responses being classified as PR. In a post hoc analysis according to PET/CT-based 2014 Lugano response assessments19 (criteria that were defined after study initiation), CMR was 80%, thus exceeding the primary response expectations, and ORR was 94%. Indeed, 13 of the aforementioned 17 patients who were classified as in PR per the 1999 IWG criteria were classified as complete metabolic responses according to PET/CT imaging.
BM involvement is a frequent observation in patients with FL, and the definition for CR or CRu according to the 1999 IWG criteria18 requires BM negativity at the end of treatment of all patients with an indeterminate, positive, or missing BM biopsy results at baseline. However, several recent studies have shown a low proportion of response reclassification based on BM assessment at the end of treatment (<5% of cases in the GALLIUM [A Study of Obinutuzumab Plus Chemotherapy in Comparison With Rituximab Plus Chemotherapy Followed by Obinutuzumab or Rituximab Maintenance in Patients With Untreated Advanced Indolent Non-Hodgkin’s Lymphoma] study and in other clinical trials).20,21 Altogether, these data largely support the evaluation of CMR according to the post hoc 2014 Lugano assessment19 in the current study.
The 80% CMR according to the 2014 Lugano response criteria in this study is in line with the high 92% CR reported by Nastoupil et al22 at first evaluation (day 1 of cycle 4) of a similar obinutuzumab and lenalidomide combination strategy in previously untreated patients with high tumor burden FL. In a subpopulation of the GALLIUM study, complete metabolic response according to the Lugano criteria was 73% (95% CI, 67-78) in the rituximab-based chemotherapy group compared with 79% (95% CI, 74-83) in the obinutuzumab-based chemotherapy group.23 CRR at 30 months [CR30] is a validated surrogate end point for PFS in FL. Here, CR30 according to the Cheson 99 criteria was 63% compared with 48% as assessed by independent review committee (55% by investigator) in the lenalidomide/rituximab arm of treatment in the RELEVANCE (A Phase 3 Open Label Randomized Study to Compare the Efficacy and Safety of Rituximab Plus Lenalidomide Versus Rituximab Plus Chemotherapy Followed by Rituximab in Subjects With Previously Untreated Follicular Lymphoma) trial.10 Although no formal comparison can be made, this strengthens the efficacy of the GALEN regimen.
After a median follow-up of 3.7 years, 3-year PFS was 82% per investigator assessment. The prolonged PFS observed in the study is consistent with the high CRR observed according to the 2014 Lugano criteria19 at the end of induction. In the rituximab plus chemotherapy followed by rituximab maintenance arms of the GALLIUM, PRIMA [Primary Rituximab and Maintenance], and RELEVANCE randomized phase 3 trials, 3-year PFS rates (by investigator assessment) were 73% (95% CI, 69-77), 75% (95% CI, 71-79), and 78% (95% CI, 74-81), respectively.10,15,24 In the rituximab plus lenalidomide arm of the RELEVANCE, CALGB (Cancer and Leukemia Group B) 50803, and M.D. Anderson Cancer Center studies, 3-year PFS rates were 77% (95% CI, 72-80), 81% (95% CI, 69-89), and 79% (95% CI, 67-92).10-12 Altogether, and within all possible pitfalls of historical, cross-trial comparisons, obinutuzumab plus lenalidomide seems to represent a promising combination strategy. In the obinutuzumab plus chemotherapy arm of the GALLIUM study, 3-year PFS was 80% (95% CI, 76-84),15 within the range of figures observed in the current study. Regarding early progression of disease, 14% of patients experienced POD24 in our study relative to 19% in the NLCS (Netherlands Cohort Study ) group after first-line R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone),25 16% after rituximab plus chemotherapy, and 10% after obinutuzumab plus chemotherapy in the GALLIUM study.26 Therefore, the GALEN combination seems to limit early progression, at least as potently as anti-CD20 plus chemotherapy. Regarding treatment duration, shorter treatment with lenalidomide and anti-CD20 has been used in the R/R setting. Both the AUGMENT (Rituximab Plus Lenalidomide for Patients With Relapsed/Refractory Indolent Non-Hodgkin's Lymphoma [Follicular Lymphoma and Marginal Zone Lymphoma]) and CALGB studies assessed the efficacy of a 12-month combination strategy.9,12 Extended duration in the MAGNIFY (Lenalidomide Plus Rituximab Followed by Lenalidomide Versus Rituximab Maintenance for Relapsed/Refractory Follicular, Marginal Zone Lymphoma) trial (#NCT01996865) for 2.5 years compared with rituximab alone will provide valuable information regarding the utility of a prolonged combination strategy in R/R FL.7 Recently reported long-term follow-up of the RELEVANCE study found that the association of rituximab and lenalidomide for the same duration as in the current study produced excellent long-term outcome, with a 6-year PFS of 60%, superimposable to the outcome yielded by rituximab plus chemotherapy.27 Whether shorter duration of anti-CD20 and lenalidomide as in R/R FL could produce similar efficacy as first-line therapy remains hypothetical.
With respect to safety, obinutuzumab plus lenalidomide was well tolerated. The most frequent grade ≥3 AE was neutropenia, which occurred in 47% of patients, with only 2 patients experiencing febrile neutropenia. No other grade ≥3 AEs occurred at a frequency >5%. Other frequent, but predominantly grade 1/2, any-grade AEs were asthenia (51%), digestive AEs (constipation, 42%; diarrhea, 35%), cough (36%), and infusion-related reaction during or after obinutuzumab infusion (28%). Although not directly comparable, the lenalidomide/rituximab arm of the RELEVANCE trial reported 32% grade ≥3 neutropenia, and any-grade fatigue, constipation, diarrhea, and infusion-related reaction in 23%, 35%, 37%, and 13% of patients, respectively.10 As expected from the known safety profile of obinutuzumab, the GALEN regimen seems associated with a slightly increased risk of grade 3/4 neutropenia and of grade 1/2 infusion-related reactions but comparable digestive grade 1/2 toxicity. Rash was similarly reported in 29% of patients in RELEVANCE receiving lenalidomide and rituximab compared with 25% of patients receiving the GALEN combination. Twelve SPMs were observed here among 11 (11%) patients. Two were deemed unrelated to the GALEN regimen because of already-associated abnormalities observed on the patients’ baseline CT imaging. In a similar setting but after a shorter median follow-up of 38 months (compared with 46 months here), second primary cancers were reported in 38 patients (7%) in the lenalidomide/rituximab group, including 25 patients who had invasive second tumors and 13 who had noninvasive second tumors. No new safety signals were therefore observed with the GALEN combination.
Results from our post hoc evaluation of response according to the 2014 Lugano criteria reiterated the importance of integrating PET/CT-based assessments to provide a comprehensive evaluation of response and to better enable cross-trial comparison with current studies. Altogether, and within the limitation of a noncontrolled, phase 2 study, the combination of obinutuzumab and lenalidomide seems to offer a promising chemotherapy-free backbone in previously untreated, high tumor burden FL to be further investigated and built upon. In particular, other agents targeting the FL microenvironment or T-cell engager allowing concomitant use of anti-CD20 monoclonal antibodies would be attractive partners for future therapeutic association with the GALEN backbone.
Acknowledgments
The authors thank all the patients, families, and caregivers who participated in the study. They also acknowledge LYSARC (The Lymphoma Academic Research Organisation) for sponsoring the trial, coordinating the study sites, and conducting the analysis and every research team and nurse in the participating centers. Writing and editorial assistance were provided by Benjamin Levine of Bio Connections LLC, funded by Bristol Myers Squibb Company.
The study was funded by LYSARC and financially supported in part by investigator-initiated study grants from Celgene/BMS and Roche.
Authorship
Contribution: E.B., R.H., and F.M. designed and performed the research and wrote the manuscript. All authors collected, analyzed, and interpreted the data.
Conflict-of-interest disclosure: E.B. declares advisory boards for Roche, Takeda, BMS, and Incyte; and honoraria for Novartis, Kite/Gilead, and Roche. P.F. reports advisory boards and travel accommodation for Roche and Celgene/BMS. F.B. declares advisory boards for BMS. H.T. declares honoraria and advisory boards for Roche. F.M. reports consultancy for Roche/Genentech; and advisory boards for Roche, BMS, Gilead, Novartis, Epizyme, Genmab, AbbVie, and Kymera. The remaining authors declare no competing financial interests.
The current affiliation for R.B. is Hôpital Privé de Provence, Aix en Provence, France.
The current affiliation for G.S. is Division of Hematologic Malignancies, Memorial Sloan Kettering Cancer Center, New York, NY.
Correspondence: Franck Morschhauser, University of Lille, CHU Lille, ULR 7365-GRITA-Groupe de Recherche sur les formes Injectables et les Technologies Associées, F-59000 Lille, France; e-mail: franck.morschhauser@chru-lille.fr.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal