

Abstract
Langerhans cell histiocytosis (LCH) can affect children and adults with a wide variety of clinical manifestations, including unifocal, single-system multifocal, single-system pulmonary (smoking-associated), or multisystem disease. The existing paradigms in the management of LCH in adults are mostly derived from the pediatric literature. Over the last decade, the discovery of clonality and MAPK-ERK pathway mutations in most cases led to the recognition of LCH as a hematopoietic neoplasm, opening the doors for treatment with targeted therapies. These advances have necessitated an update of the existing recommendations for the diagnosis and treatment of LCH in adults. This document presents consensus recommendations that resulted from the discussions at the annual Histiocyte Society meeting in 2019, encompassing clinical features, classification, diagnostic criteria, treatment algorithm, and response assessment for adults with LCH. The recommendations favor the use of 18F-Fluorodeoxyglucose positron emission tomography-based imaging for staging and response assessment in the majority of cases. Most adults with unifocal disease may be cured by local therapies, while the first-line treatment for single-system pulmonary LCH remains smoking cessation. Among patients not amenable or unresponsive to these treatments and/or have multifocal and multisystem disease, systemic treatments are recommended. Preferred systemic treatments in adults with LCH include cladribine or cytarabine, with the emerging role of targeted (BRAF and MEK inhibitor) therapies. Despite documented responses to treatments, many patients struggle with a high symptom burden from pain, fatigue, and mood disorders that should be acknowledged and managed appropriately.
Introduction
Langerhans cell histiocytosis (LCH) is a rare hematologic disorder that affects children and adults. LCH was one of the first histiocytic disorders to be recognized as a hematopoietic neoplasm by the World Health Organization (WHO) due to the establishment of clonality.1 Subsequently, the discovery of BRAF-V600E mutation in about 50% of cases solidified the notion that LCH is a neoplastic disease.2 LCH is included among the “L-group” of the revised classification of histiocytic disorders and is grouped along with Erdheim-Chester disease (ECD).3 Most of our current understanding of LCH is derived from pediatric studies, with a paucity of data examining adult counterparts. The exact incidence of adult LCH is undefined, estimated at 1 to 1.5 cases per million population per year.4,5 Since the previous publication of adult LCH guidelines in 2013,6 several key advances have occurred in our understanding of the disease, including the discovery of MAPK pathway (RAS-RAF-MEK-ERK) mutations beyond BRAF-V600E7,8 and efficacious treatments using kinase pathway inhibitors.9,10 Tissue next-generation sequencing (NGS) studies have revealed that BRAF- wild-type LCH may harbor several other mutations in the MAPK-ERK pathway genes (eg, MAP2K1, MAP3K1, ARAF, NRAS, KRAS).7 Similar to ECD, kinase fusions (BRAF, ALK, NTRK1), BRAF- duplications, insertions, deletions, and rare mutations in the PI3K-AKT-mTOR pathway have been reported in LCH as well.7,11,12 These molecular discoveries have implications for the diagnosis and management of LCH, and in conjunction with other recent studies on LCH in adults, have necessitated an update of the existing recommendations.
Methods
An international, multidisciplinary group of physicians and scientists convened at the annual Histiocyte Society meeting on 3 November 2019 to initiate the process of updating the adult LCH consensus recommendations. The group was comprised of experts from multiple subspecialties with experience in the clinical care/research of LCH and related histiocytic disorders (supplemental Table 1). The proposed update was based on 1) novel data since prior guideline publication in 2013 and 2) group members’ evolving experience with LCH in adults. We generated key recommendation statements (consensus statements or CS#1-32) (Table 1) and recorded the votes by coauthors as “agreement,” “disagreement,” and “not-qualified to answer” using a modified Delphi approach as previously described (supplemental Appendix 1).13 The degree of consensus for each statement was categorized into: A (strong consensus: ≥95%), B (consensus: 75-94%), and C (majority agreement: 50-74%).
Consensus recommendations for the diagnosis and management of adult LCH
| Statement number | Consensus statements | Consensus recommendation category |
|---|---|---|
| Diagnosis | ||
| 1. | A biopsy of lesional tissue is recommended even in circumstances of highly suggestive clinical and imaging features to confirm LCH diagnosis and establish BRAF or another MAPK-ERK pathway mutational status. Cases of single-system PLCH with typical radiologic findings and clinical context are a reasonable exception, although a biopsy is recommended in these cases as well. | A |
| 2. | LCH should be considered in the presence of characteristic clinical/radiologic features (Table 3), even when a histopathologic review is equivocal. Molecular analysis of tissue for BRAF and MAPK-ERK pathway mutations can be helpful in the diagnosis of questionable lesions. | B |
| 3. | Baseline full-body (vertex-to-toes) FDG-PET/CT, including the distal extremities, is recommended to aid in diagnosis and defining the extent of disease. | B |
| 4. | Organ-specific imaging (CT, MRI) is recommended to further assess involved sites of disease based on initial imaging studies. | A |
| 5. | MRI of the brain with gadolinium, with a dedicated examination of the sella turcica, should be undertaken at diagnosis in cases with pituitary dysfunction or neurologic symptoms. | A |
| 6. | In patients with suspected/confirmed PLCH, HRCT of the chest should be performed. | A |
| 7. | In patients with single-system PLCH, a surgical lung biopsy may be necessary to confirm the diagnosis if a bronchoscopic biopsy or other methods are nondiagnostic. | A |
| 8. | All patients with PLCH should undergo pulmonary function testing (spirometry with lung volumes, diffusion capacity, and plethysmography) at the time of diagnosis. | A |
| 9. | All patients with PLCH who are symptomatic or have an abnormal diffusing capacity for carbon monoxide should undergo a resting transthoracic echocardiogram to screen for pulmonary hypertension. | B |
| 10. | Right-sided heart catheterization and vasoreactivity testing should be considered in selected patients with echocardiographically demonstrated pulmonary hypertension to assess its severity and aid with further management. | B |
| 11. | MRCP or ERCP should be performed in cases with elevated serum cholestasis markers or sonomorphologically-dilated bile ducts to evaluate for sclerosing cholangitis related to LCH. | A |
| 12. | For patients with suspected sclerosing cholangitis as a manifestation of LCH, early liver biopsy should be performed for histopathologic and mutational assessment. | A |
| 13. | Laboratory studies to assess for liver insufficiency, cytopenias, markers of inflammation (C-reactive protein) should be performed at diagnosis. | A |
| 14. | For patients with polyuria/polydipsia or involvement of pituitary/hypothalamus axis on cranial imaging, laboratory evaluation should be undertaken to rule out DI and anterior pituitary function. | A |
| 15. | Currently, there is no role for routine bone marrow biopsy in adult LCH. However, due to a high prevalence of concomitant and subsequent myeloid neoplasms in patients with LCH, bone marrow biopsy should be considered in the context of otherwise unexplained cytopenias or cytosis. | A |
| 16. | All patients with LCH should undergo BRAF-V600E mutational testing to aid in diagnosis and treatment. | A |
| 17. | IHC for VE1 may not be a sensitive or specific marker for BRAF-V600E mutational analysis and should be confirmed with another molecular assay if feasible. | B |
| 18. | For BRAF-V600-wt LCH cases, next-generation sequencing should be undertaken to assess for MAPK-ERK pathway mutations, especially in situations where the diagnosis is questionable or second-line treatment is needed. | A |
| 19. | In the absence of sufficient tumor tissue, cell-free DNA analysis from peripheral blood can be used for the assessment of BRAF-mutational status. However, the sensitivity of such assays may be variable. | A |
| Treatment | Unifocal LCH | |
| 20. | For unifocal LCH (except DI), observation or local therapies such as surgical excision, intralesional steroids, or radiation are recommended as first-line treatments. | B |
| 21. | For unifocal LCH involving specific sites (nervous system, liver, spleen, etc), systemic treatment should be implemented. | A |
| 22. | For unifocal LCH of pituitary/hypothalamus resulting in DI and anterior pituitary dysfunction, hormone replacement should be undertaken. The role of systemic therapy is unclear and is recommended in cases with symptoms that are recent-onset or when a radiologic lesion is present. | B |
| Single-system pulmonary LCH | ||
| 23. | Cessation of smoking, vaping, inhalation of marijuana or other substances is recommended as first-line therapy for single-system PLCH. | A |
| 24. | Systemic therapy is recommended for single-system PLCH in the presence of progressive disease (regardless of smoking status) or for stable disease with clinically significant respiratory symptoms or dysfunction. | A |
| 25. | For patients who develop advanced single-system PLCH refractory to or ineligible for systemic treatments, lung transplantation referral should be undertaken. | A |
| Multifocal and multisystem LCH | ||
| 26. | For multifocal osseous LCH, recommended treatments are radiation therapy (<3 lesions safely amenable to radiation), bisphosphonates, or systemic chemotherapy. | B |
| 27. | For multifocal cutaneous LCH, recommended treatments are topical therapy, oral low-dose weekly methotrexate ± prednisone/6-MP, hydroxyurea, or IMiDs. | B |
| 28. | For multisystem LCH or extensive/refractory multifocal single-system LCH, systemic chemotherapy agents such as cladribine, cytarabine, or vinblastine + prednisone are recommended. | B |
| 29. | For LCH involving the brain parenchyma, first-line treatment with chemotherapy with cladribine or cytarabine is recommended. | A |
| 30. | For LCH refractory to first-line treatment or with end-organ dysfunction (e.g., neurologic impairment, sclerosing cholangitis), alternate conventional treatment or targeted therapies (BRAF or MEK inhibitors) should be implemented. | A |
| Response assessment and monitoring | ||
| 31. | The type and frequency of response assessments and follow-up examinations are variable and dependent on the degree of involvement with LCH (Table 7). | A |
| 32. | For initially FDG PET avid LCH, it is recommended to repeat an FDG PET-based imaging study for assessment of disease response after 2-3 mo of initiation of therapy, with subsequent imaging frequency tailored individually based on the specific clinical scenario. | A |
| Statement number | Consensus statements | Consensus recommendation category |
|---|---|---|
| Diagnosis | ||
| 1. | A biopsy of lesional tissue is recommended even in circumstances of highly suggestive clinical and imaging features to confirm LCH diagnosis and establish BRAF or another MAPK-ERK pathway mutational status. Cases of single-system PLCH with typical radiologic findings and clinical context are a reasonable exception, although a biopsy is recommended in these cases as well. | A |
| 2. | LCH should be considered in the presence of characteristic clinical/radiologic features (Table 3), even when a histopathologic review is equivocal. Molecular analysis of tissue for BRAF and MAPK-ERK pathway mutations can be helpful in the diagnosis of questionable lesions. | B |
| 3. | Baseline full-body (vertex-to-toes) FDG-PET/CT, including the distal extremities, is recommended to aid in diagnosis and defining the extent of disease. | B |
| 4. | Organ-specific imaging (CT, MRI) is recommended to further assess involved sites of disease based on initial imaging studies. | A |
| 5. | MRI of the brain with gadolinium, with a dedicated examination of the sella turcica, should be undertaken at diagnosis in cases with pituitary dysfunction or neurologic symptoms. | A |
| 6. | In patients with suspected/confirmed PLCH, HRCT of the chest should be performed. | A |
| 7. | In patients with single-system PLCH, a surgical lung biopsy may be necessary to confirm the diagnosis if a bronchoscopic biopsy or other methods are nondiagnostic. | A |
| 8. | All patients with PLCH should undergo pulmonary function testing (spirometry with lung volumes, diffusion capacity, and plethysmography) at the time of diagnosis. | A |
| 9. | All patients with PLCH who are symptomatic or have an abnormal diffusing capacity for carbon monoxide should undergo a resting transthoracic echocardiogram to screen for pulmonary hypertension. | B |
| 10. | Right-sided heart catheterization and vasoreactivity testing should be considered in selected patients with echocardiographically demonstrated pulmonary hypertension to assess its severity and aid with further management. | B |
| 11. | MRCP or ERCP should be performed in cases with elevated serum cholestasis markers or sonomorphologically-dilated bile ducts to evaluate for sclerosing cholangitis related to LCH. | A |
| 12. | For patients with suspected sclerosing cholangitis as a manifestation of LCH, early liver biopsy should be performed for histopathologic and mutational assessment. | A |
| 13. | Laboratory studies to assess for liver insufficiency, cytopenias, markers of inflammation (C-reactive protein) should be performed at diagnosis. | A |
| 14. | For patients with polyuria/polydipsia or involvement of pituitary/hypothalamus axis on cranial imaging, laboratory evaluation should be undertaken to rule out DI and anterior pituitary function. | A |
| 15. | Currently, there is no role for routine bone marrow biopsy in adult LCH. However, due to a high prevalence of concomitant and subsequent myeloid neoplasms in patients with LCH, bone marrow biopsy should be considered in the context of otherwise unexplained cytopenias or cytosis. | A |
| 16. | All patients with LCH should undergo BRAF-V600E mutational testing to aid in diagnosis and treatment. | A |
| 17. | IHC for VE1 may not be a sensitive or specific marker for BRAF-V600E mutational analysis and should be confirmed with another molecular assay if feasible. | B |
| 18. | For BRAF-V600-wt LCH cases, next-generation sequencing should be undertaken to assess for MAPK-ERK pathway mutations, especially in situations where the diagnosis is questionable or second-line treatment is needed. | A |
| 19. | In the absence of sufficient tumor tissue, cell-free DNA analysis from peripheral blood can be used for the assessment of BRAF-mutational status. However, the sensitivity of such assays may be variable. | A |
| Treatment | Unifocal LCH | |
| 20. | For unifocal LCH (except DI), observation or local therapies such as surgical excision, intralesional steroids, or radiation are recommended as first-line treatments. | B |
| 21. | For unifocal LCH involving specific sites (nervous system, liver, spleen, etc), systemic treatment should be implemented. | A |
| 22. | For unifocal LCH of pituitary/hypothalamus resulting in DI and anterior pituitary dysfunction, hormone replacement should be undertaken. The role of systemic therapy is unclear and is recommended in cases with symptoms that are recent-onset or when a radiologic lesion is present. | B |
| Single-system pulmonary LCH | ||
| 23. | Cessation of smoking, vaping, inhalation of marijuana or other substances is recommended as first-line therapy for single-system PLCH. | A |
| 24. | Systemic therapy is recommended for single-system PLCH in the presence of progressive disease (regardless of smoking status) or for stable disease with clinically significant respiratory symptoms or dysfunction. | A |
| 25. | For patients who develop advanced single-system PLCH refractory to or ineligible for systemic treatments, lung transplantation referral should be undertaken. | A |
| Multifocal and multisystem LCH | ||
| 26. | For multifocal osseous LCH, recommended treatments are radiation therapy (<3 lesions safely amenable to radiation), bisphosphonates, or systemic chemotherapy. | B |
| 27. | For multifocal cutaneous LCH, recommended treatments are topical therapy, oral low-dose weekly methotrexate ± prednisone/6-MP, hydroxyurea, or IMiDs. | B |
| 28. | For multisystem LCH or extensive/refractory multifocal single-system LCH, systemic chemotherapy agents such as cladribine, cytarabine, or vinblastine + prednisone are recommended. | B |
| 29. | For LCH involving the brain parenchyma, first-line treatment with chemotherapy with cladribine or cytarabine is recommended. | A |
| 30. | For LCH refractory to first-line treatment or with end-organ dysfunction (e.g., neurologic impairment, sclerosing cholangitis), alternate conventional treatment or targeted therapies (BRAF or MEK inhibitors) should be implemented. | A |
| Response assessment and monitoring | ||
| 31. | The type and frequency of response assessments and follow-up examinations are variable and dependent on the degree of involvement with LCH (Table 7). | A |
| 32. | For initially FDG PET avid LCH, it is recommended to repeat an FDG PET-based imaging study for assessment of disease response after 2-3 mo of initiation of therapy, with subsequent imaging frequency tailored individually based on the specific clinical scenario. | A |
A (strong consensus: ≥95%), B (consensus: 75-94%), and C (majority agreement: 50-74%).
6-MP, 6-mercaptopurine; CNS, central nervous system; CT, computed tomography; DI, diabetes insipidus; ERCP, endoscopic retrograde cholangiopancreatography; FDG-PET, 18Fluorodeoxyglucose positron emission tomography; IMiDs, immunomodulators (thalidomide, lenalidomide); MRCP, magnetic resonance cholangiopancreatography; MRI, magnetic resonance imaging.
Pulmonary LCH—a distinct entity?
Pulmonary LCH (PLCH) may occur as part of multisystem LCH or, more commonly, as an isolated disease (single-system PLCH).13 Single-system PLCH was initially thought to be an inflammatory/reactive disorder given the near-universal association with smoking14 and instances of remission upon smoking cessation.15 Earlier studies assessing clonality of single-system PLCH showed mixed results,16,17 but more recent studies have identified recurrent MAPK pathway alterations in 85% of these lesions, including BRAF-V600E in 36% to 50% of cases.18,19 The latter finding supports the contention that single-system PLCH is indeed a clonal process; more studies are needed to understand its natural history and the role of smoking in disease modulation.20
LCH subtypes and definitions
Given the significant variability in clinical manifestations, various attempts have been undertaken to classify LCH in pediatric and adult populations.3,21 To harmonize the definitions in light of recent evidence in adults with LCH, we propose the classification outlined in Table 2. It should be noted that our classification differs from the 2016 Histiocyte Society classification3 in that we do not differentiate risk-organ disease as a separate entity due to a lack of data on prognostic and therapeutic implications in adults in the era of targeted therapies. The proposed classification system differentiates between unifocal (solitary lesion) and single-system multifocal disease (>1 lesion). In multisystem LCH, ≥2 organs/organ systems are involved. We also propose a distinct category of single-system PLCH due to its unique etiopathogenesis and treatment. PLCH with extrapulmonary disease is classified under multisystem disease. Every attempt should be undertaken to appropriately define the disease extent using radiographic studies mentioned in the baseline evaluations section below.
Classification of LCH in adults
| Subtype | Definition |
|---|---|
| Unifocal | Solitary lesion involving any organ |
| Single-system pulmonary | Isolated lung involvement (predominantly smoking related) |
| Single-system multifocal | >1 lesion involving any organ |
| Multisystem | ≥2 organ/system involvement |
| Subtype | Definition |
|---|---|
| Unifocal | Solitary lesion involving any organ |
| Single-system pulmonary | Isolated lung involvement (predominantly smoking related) |
| Single-system multifocal | >1 lesion involving any organ |
| Multisystem | ≥2 organ/system involvement |
Mixed histiocytosis
Although LCH has distinct clinical and morphologic characteristics compared with ECD and Rosai-Dorfman disease (RDD), several reports of coexistence of these disorders as “mixed” or “overlap” histiocytosis exist. A case series noted LCH-like lesions cooccurring with RDD, most commonly in young patients and involving lymph nodes.22 In 90% of these instances, LCH constituted a minor component of the lymph node sections (<10%) compared with RDD. LCH/ECD overlap was reported in a French series of 23 patients, where the ECD component was found to occur either subsequently or concomitantly with LCH but never before it.23 In contrast with the LCH/RDD overlap described earlier, the LCH and ECD lesions occurred less frequently in the same anatomic site (26%). These cases had a high frequency of BRAF-V600E mutations in the LCH (69%) and ECD (82%) lesions.23 Pediatric reports have suggested that LCH may also occur concomitantly with juvenile xanthogranuloma.24 These data highlight that the histiocytic disorders may potentially lie on a clinicopathologic spectrum; cognizance of the mixed histiocytoses may warrant further investigations, such as additional biopsies, especially at recurrence or unusual disease sites in order to diagnose these entities and tailor their management.
LCH and concomitant hematologic/solid neoplasms
Several institutional and population-based studies have shown high incidence and prevalence of other malignancies in patients with LCH, including myeloid neoplasms (acute myeloid leukemia,4 myeloproliferative neoplasms, chronic myelomonocytic leukemia,25,26 etc), lymphomas, and solid organ malignancies (especially lung and thyroid cancer).27-31 LCH of the thyroid gland can cooccur with papillary thyroid cancer harboring BRAF-V600E mutations.29,32 LCH foci have also been found incidentally around resected specimens of renal cell carcinoma.30 Patients with PLCH, in particular, have a high incidence of concomitant lung carcinomas, likely due to their smoking history.33 LCH-like lesions and aggregates of Langerhans cells (LCs) may cooccur in lymph node biopsies of lymphomas and likely represent a “bystander” phenomenon where the lymphoma subtype drives treatment.34-36 Clinicians should be aware of the occurrence of second malignancies at any time point (before, during, or after) in relation to the LCH diagnosis as this may influence surveillance and management.
Clinical and radiographic features of LCH in adults
LCH has diverse manifestations in adults, with varying frequencies of disease sites based mostly upon institutional retrospective series (Figure 1; Table 3; supplemental Appendix 2).6,15,33,37-81 Clinicians should be familiar with the clinical and radiographic features of LCH and attempt to pursue comprehensive evaluation at baseline given the implications for diagnosis and management of the disease.
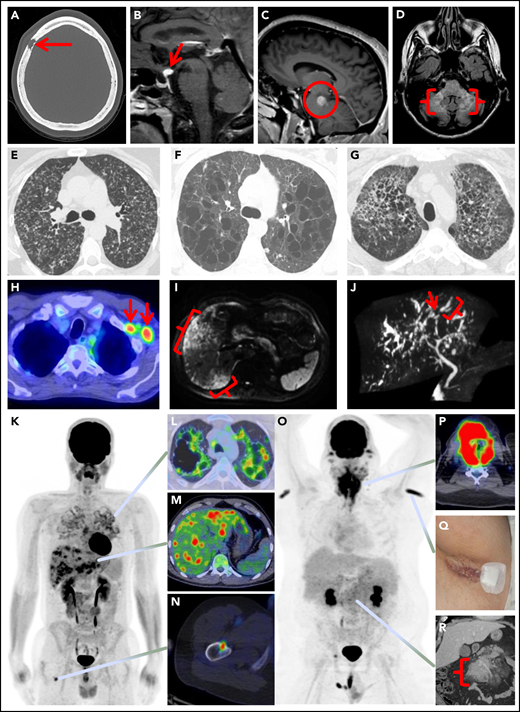
Spectrum of manifestations of LCH in adults. (A) Axial computed tomography (CT) of the calvarium demonstrating a lytic lesion with a beveled edge (arrow). (B) Sagittal pituitary T2 weighted fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging (MRI) highlighting a hypothalamic lesion with increased FLAIR signal (arrow). (C) Sagittal T1 weighted contrast-enhanced brain MRI demonstrating an enhancing posterior midbrain/pons lesion (circle). (D) Axial brain MRI with a neurodegenerative pattern of T2 signal abnormality throughout the bilateral cerebellar peduncles (brackets). (E-G) Axial CT of the mid to upper chest demonstrating variable appearances of pulmonary LCH to include the most common nodulocystic pattern ground-glass nodules and cysts (E), a cystic-predominant pattern with larger irregular cysts and a few scattered nodules (F), and more confluent combined cystic and nodular disease with architectural distortion consistent with elements of fibrosis (G). (H) Axial fused 18F-fluorodeoxyglucose positron emission tomography CT (FDG PET/CT) of the chest demonstrating 2 FDG avid left axillary lymph nodes (arrows). (I) Diffusion-weighted MRI of the liver demonstrating an infiltrative pattern of increased signal throughout the right hepatic lobe (brackets). (J) Magnetic resonance cholangiopancreatography demonstrating multifocal stricturing (arrow) and dilatation (bracket) of the intrahepatic ducts. (K) Maximum intensity projection FDG PET/CT demonstrating FDG avid advanced pulmonary disease (L), multifocal FDG avid hepatic disease (M), and an FDG avid lytic right femur lesion (N). (O) MIP FDG PET/CT demonstrating an infiltrative FDG avid laryngeal mass (P), bilateral axillary dermal lesions with associated left axillary photograph (Q), and a mildly FDG avid mesenteric mass highlighted with a bracket (R).
Spectrum of manifestations of LCH in adults. (A) Axial computed tomography (CT) of the calvarium demonstrating a lytic lesion with a beveled edge (arrow). (B) Sagittal pituitary T2 weighted fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging (MRI) highlighting a hypothalamic lesion with increased FLAIR signal (arrow). (C) Sagittal T1 weighted contrast-enhanced brain MRI demonstrating an enhancing posterior midbrain/pons lesion (circle). (D) Axial brain MRI with a neurodegenerative pattern of T2 signal abnormality throughout the bilateral cerebellar peduncles (brackets). (E-G) Axial CT of the mid to upper chest demonstrating variable appearances of pulmonary LCH to include the most common nodulocystic pattern ground-glass nodules and cysts (E), a cystic-predominant pattern with larger irregular cysts and a few scattered nodules (F), and more confluent combined cystic and nodular disease with architectural distortion consistent with elements of fibrosis (G). (H) Axial fused 18F-fluorodeoxyglucose positron emission tomography CT (FDG PET/CT) of the chest demonstrating 2 FDG avid left axillary lymph nodes (arrows). (I) Diffusion-weighted MRI of the liver demonstrating an infiltrative pattern of increased signal throughout the right hepatic lobe (brackets). (J) Magnetic resonance cholangiopancreatography demonstrating multifocal stricturing (arrow) and dilatation (bracket) of the intrahepatic ducts. (K) Maximum intensity projection FDG PET/CT demonstrating FDG avid advanced pulmonary disease (L), multifocal FDG avid hepatic disease (M), and an FDG avid lytic right femur lesion (N). (O) MIP FDG PET/CT demonstrating an infiltrative FDG avid laryngeal mass (P), bilateral axillary dermal lesions with associated left axillary photograph (Q), and a mildly FDG avid mesenteric mass highlighted with a bracket (R).
Summary of organ manifestations of LCH in adults
| Organ/organ system | Clinical and radiographic features |
|---|---|
| Bones | Lytic osseous lesions seen in 30-50% of cases may occur as single-system or multisystem disease.15,41 Frequently involves the skull and dental sites; other sites include the pelvis, vertebrae, ribs, and extremities.51,5518F-fluorodeoxyglucose (FDG) positron emission tomography-computed tomography (FDG-PET/CT) is highly sensitive for bone lesion detection and is superior to other imaging studies like CT and MRI.38,68,69 On FDG-PET/CT, LCH bone lesions are lytic, involve the cortex, and have a “punched-out” appearance. |
| Skin | Isolated skin involvement with LCH in adults is uncommon (5-10%) and tends to occur more commonly as part of multisystem disease (20-50%).41,44 Most commonly presents as an erythematous papular rash with scaling and crusting, located on the chest, back, abdomen, limbs, scalp, or groin. Rarely can involve mucosa of the oral cavity, genitalia, or perianal region.40,47,53 |
| Endocrine | Most commonly presents as DI (25%) and may precede LCH diagnosis by years.54,65 Anterior pituitary dysfunction can be seen in ∼20% of cases.61,62 Pituitary endocrinopathies can occur without corresponding abnormal imaging studies.61,63,66,75 Most endocrinopathies may not be reversible despite systemic LCH treatments.54 |
| Nervous system and orbit | Seen in 15-20% of cases15; can occur as focal mass lesions in the pituitary stalk, hypothalamus, or pineal gland. Infiltration of the brain parenchyma is rare, most often in the cerebellum or brainstem, leading to ataxia and dysarthria.46,48 Neurodegenerative LCH is a rare manifestation with 2 components: 1) LCH-associated abnormal CNS imaging (LACI) with T2 hyperintense lesions and atrophy predominantly involving the posterior fossa,81 and 2) LCH-associated abnormal CNS symptoms (LACS) manifesting as neuropsychological impairment and gait ataxia.60 There may be a lag of 10-20 y between the diagnosis of LCH and LACI/LACS.60 Orbit involvement is very rare.49 |
| Pulmonary | PLCH can occur in 40-50% of cases, mostly presenting as single-system disease; 60-70% have respiratory symptoms, 10% may present as pneumothorax.33 Decreased diffusing capacity for carbon monoxide is the most common abnormality (80-90%) in combination with changes of either obstructive, restrictive, or mixed physiologic abnormalities; lung function tests may be normal in 10% of cases.33 Standard chest radiographic features of PLCH include upper and middle field predominant reticulonodular changes, and HRCT of the chest may show nodular or cystic lesions (Figure 1). In advanced stages of disease, cystic lesions predominate. These findings on HRCT are diagnostic of PLCH in a smoker; however, every attempt should be made for tissue acquisition unless considered unsafe. Bronchoscopy may be normal or show inflammatory changes. Bronchoalveolar lavage is rarely diagnostic of PLCH.80 PLCH lung nodules can be hypermetabolic on FDG PET-CT, which does not allow differentiation from other malignant lesions. Thick-walled cysts may also demonstrate FDG uptake.58,68 |
| Liver and spleen | Liver involvement can occur in 10-15% of cases,41 seen more commonly in multisystem LCH.37 May manifest in 2 forms: 1) early-stage disease with parenchymal infiltration (hepatomegaly, tumorous nodules, mild cholestasis) and 2) late-stage sclerosing cholangitis-like disease (severe cholestasis) that can quickly progress to end-stage liver failure and be lethal.50 Magnetic resonance cholangiopancreatography may show irregularities of intrahepatic bile ducts and is the recommended initial test (Figure 1).50 Liver biopsy may be negative for LCH and may show fibrosis or nonspecific inflammation. Spleen involvement occurs in 10-15% of patients and may coexist with liver disease.44,48 |
| Bone marrow and lymph nodes | Bone marrow involvement by LCH can occur, but the frequency is unknown.56 Presence of peripheral blood count abnormalities may point toward bone marrow infiltration by LCH or another myeloid neoplasm. Isolated LCH of lymph nodes is uncommon,72 although it can occur as part of multisystem disease in 10-30% of cases.6,15,44 |
| Gastrointestinal and cardiovascular | Gastrointestinal involvement is rare and may present with diarrhea, abdominal discomfort, or an incidental polyp on colonoscopy.43,71,77 Although the encasement of arterial structures has been reported,45 cardiovascular involvement is very rare and raises suspicion for an LCH/ECD overlap. |
| Organ/organ system | Clinical and radiographic features |
|---|---|
| Bones | Lytic osseous lesions seen in 30-50% of cases may occur as single-system or multisystem disease.15,41 Frequently involves the skull and dental sites; other sites include the pelvis, vertebrae, ribs, and extremities.51,5518F-fluorodeoxyglucose (FDG) positron emission tomography-computed tomography (FDG-PET/CT) is highly sensitive for bone lesion detection and is superior to other imaging studies like CT and MRI.38,68,69 On FDG-PET/CT, LCH bone lesions are lytic, involve the cortex, and have a “punched-out” appearance. |
| Skin | Isolated skin involvement with LCH in adults is uncommon (5-10%) and tends to occur more commonly as part of multisystem disease (20-50%).41,44 Most commonly presents as an erythematous papular rash with scaling and crusting, located on the chest, back, abdomen, limbs, scalp, or groin. Rarely can involve mucosa of the oral cavity, genitalia, or perianal region.40,47,53 |
| Endocrine | Most commonly presents as DI (25%) and may precede LCH diagnosis by years.54,65 Anterior pituitary dysfunction can be seen in ∼20% of cases.61,62 Pituitary endocrinopathies can occur without corresponding abnormal imaging studies.61,63,66,75 Most endocrinopathies may not be reversible despite systemic LCH treatments.54 |
| Nervous system and orbit | Seen in 15-20% of cases15; can occur as focal mass lesions in the pituitary stalk, hypothalamus, or pineal gland. Infiltration of the brain parenchyma is rare, most often in the cerebellum or brainstem, leading to ataxia and dysarthria.46,48 Neurodegenerative LCH is a rare manifestation with 2 components: 1) LCH-associated abnormal CNS imaging (LACI) with T2 hyperintense lesions and atrophy predominantly involving the posterior fossa,81 and 2) LCH-associated abnormal CNS symptoms (LACS) manifesting as neuropsychological impairment and gait ataxia.60 There may be a lag of 10-20 y between the diagnosis of LCH and LACI/LACS.60 Orbit involvement is very rare.49 |
| Pulmonary | PLCH can occur in 40-50% of cases, mostly presenting as single-system disease; 60-70% have respiratory symptoms, 10% may present as pneumothorax.33 Decreased diffusing capacity for carbon monoxide is the most common abnormality (80-90%) in combination with changes of either obstructive, restrictive, or mixed physiologic abnormalities; lung function tests may be normal in 10% of cases.33 Standard chest radiographic features of PLCH include upper and middle field predominant reticulonodular changes, and HRCT of the chest may show nodular or cystic lesions (Figure 1). In advanced stages of disease, cystic lesions predominate. These findings on HRCT are diagnostic of PLCH in a smoker; however, every attempt should be made for tissue acquisition unless considered unsafe. Bronchoscopy may be normal or show inflammatory changes. Bronchoalveolar lavage is rarely diagnostic of PLCH.80 PLCH lung nodules can be hypermetabolic on FDG PET-CT, which does not allow differentiation from other malignant lesions. Thick-walled cysts may also demonstrate FDG uptake.58,68 |
| Liver and spleen | Liver involvement can occur in 10-15% of cases,41 seen more commonly in multisystem LCH.37 May manifest in 2 forms: 1) early-stage disease with parenchymal infiltration (hepatomegaly, tumorous nodules, mild cholestasis) and 2) late-stage sclerosing cholangitis-like disease (severe cholestasis) that can quickly progress to end-stage liver failure and be lethal.50 Magnetic resonance cholangiopancreatography may show irregularities of intrahepatic bile ducts and is the recommended initial test (Figure 1).50 Liver biopsy may be negative for LCH and may show fibrosis or nonspecific inflammation. Spleen involvement occurs in 10-15% of patients and may coexist with liver disease.44,48 |
| Bone marrow and lymph nodes | Bone marrow involvement by LCH can occur, but the frequency is unknown.56 Presence of peripheral blood count abnormalities may point toward bone marrow infiltration by LCH or another myeloid neoplasm. Isolated LCH of lymph nodes is uncommon,72 although it can occur as part of multisystem disease in 10-30% of cases.6,15,44 |
| Gastrointestinal and cardiovascular | Gastrointestinal involvement is rare and may present with diarrhea, abdominal discomfort, or an incidental polyp on colonoscopy.43,71,77 Although the encasement of arterial structures has been reported,45 cardiovascular involvement is very rare and raises suspicion for an LCH/ECD overlap. |
Diagnosis of LCH in adults
Diagnostic approach
The diagnosis of LCH usually relies on a histopathologic review of lesional tissue. All attempts should be made to obtain a tissue biopsy, not only to achieve a diagnosis but also for mutational analysis that can direct therapy; however, in instances without classic pathologic findings or inaccessible/insufficient tissue specimen, a presumptive diagnosis can be made based on characteristic clinical, radiographic, or molecular features (Table 4; CS#1-2). Definitive diagnosis of single-system PLCH may be obtained through transbronchial, transthoracic, or, more commonly, by surgical lung biopsy.82,83 In cases where a lung biopsy is not considered safe, a diagnosis of single-system PLCH can be rendered in the presence of typical radiologic findings on high-resolution computed tomography (HRCT) of the chest in a smoker after careful ruling out of other etiologies (Table 4; CS#1).83,84 In such cases, circulating DNA analysis for MAPK-ERK mutations may help solidify the diagnosis. Among patients who have undergone FDG-PET/CT, the safely accessible sites of greatest FDG hypermetabolism should be targeted for biopsy. We recommend acquiring multiple (5-10) core biopsies (16-18 gauge) instead of fine-needle aspiration, given the variable cellularity of lesions. Bone specimens present unique challenges, including crushed samples and interference with mutational analysis from decalcification, including false-negative immunohistochemistry (IHC) results for BRAF-V600E. To circumvent this issue, it is recommended to either preserve a fragment of tissue without acid decalcification or use ethylenediaminetetraacetic acid (EDTA)-based decalcification to maintain DNA integrity.
Characteristic features of LCH in adults
| Clinical and radiographic features |
| 1. Upper lobe predominant nodular and cystic lung lesions in a smoker |
| 2. Central diabetes insipidus |
| 3. Punched-out lytic osseous lesions, often involving flat bones (skull, sternum, ribs, pelvis) |
| Histopathologic features |
| 1. Lesional histiocytes with elongated, grooved nuclei, often with intermixed eosinophils |
| 2. CD207 (langerin)- and CD1a-positive histiocytes in lesional tissue by IHC |
| 3. Characteristic pattern of tissue involvement to exclude reactive Langerhans cells |
| Molecular features |
| 1. BRAF-V600E mutation |
| 2. Other activating mutations in the RAS-RAF-MEK-ERK pathway (MAP2K1, BRAF, KRAS, NRAS, ARAF, etc) |
| 3. Activating kinase fusions |
| Clinical and radiographic features |
| 1. Upper lobe predominant nodular and cystic lung lesions in a smoker |
| 2. Central diabetes insipidus |
| 3. Punched-out lytic osseous lesions, often involving flat bones (skull, sternum, ribs, pelvis) |
| Histopathologic features |
| 1. Lesional histiocytes with elongated, grooved nuclei, often with intermixed eosinophils |
| 2. CD207 (langerin)- and CD1a-positive histiocytes in lesional tissue by IHC |
| 3. Characteristic pattern of tissue involvement to exclude reactive Langerhans cells |
| Molecular features |
| 1. BRAF-V600E mutation |
| 2. Other activating mutations in the RAS-RAF-MEK-ERK pathway (MAP2K1, BRAF, KRAS, NRAS, ARAF, etc) |
| 3. Activating kinase fusions |
Histopathologic features and differential diagnosis
On tissue biopsy, the LCH infiltrate is characterized by clusters of intermediate-sized cells with reniform nuclei, frequent longitudinal nuclear grooves, dispersed chromatin, and abundant eosinophilic cytoplasm.3,48,70 Occasional multinucleated giant cells may be present. A reactive inflammatory cell background is common and includes eosinophils, small lymphocytes, and macrophages (Figure 2). In PLCH, the neoplastic LCs are preferentially located in and destroy the walls of distal bronchioles.84 In certain anatomic sites, in particular bone and lung, there is an evolution of the lesions over time from cellular infiltrates to “burned-out” lesions with patchy cellularity, increased fibrosis, and foamy histiocytes.3,85 Within skin lesions, the neoplastic cells of LCH are distinguished from reactive LC populations by their enlarged nuclei, sheet-like collections with involvement of the epidermis, and lack of dendritic processes.86 In lymph nodes, LCH involves and expands the sinuses, with their microanatomic location being a critical distinction from the reactive LCs that are present in the paracortex of lymph nodes with dermatopathic change.72
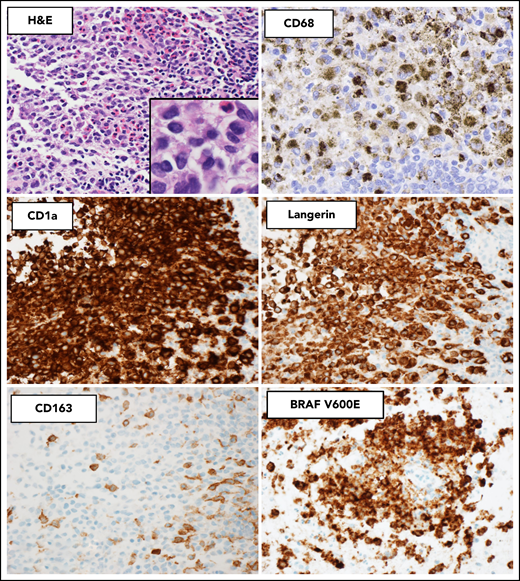
Histopathologic and immunophenotypic features of LCH. The characteristic infiltrate of LCH on hematoxylin and eosin shows mononuclear cells with grooved nuclei and abundant eosinophilic cytoplasm, with a mixture of eosinophils and small lymphocytes. The LCH cells show expression of CD68 (cytoplasmic, Golgi dot-like staining with background macrophages darkly stained), CD1a (surface), and langerin (cytoplasmic) and are negative for CD163. The mutant-specific antibody clone VE1 detects the BRAF-V600E mutation by immunohistochemistry with 2-3+ strong cytoplasmic staining. Images magnification ×400; inset magnification ×600.
Histopathologic and immunophenotypic features of LCH. The characteristic infiltrate of LCH on hematoxylin and eosin shows mononuclear cells with grooved nuclei and abundant eosinophilic cytoplasm, with a mixture of eosinophils and small lymphocytes. The LCH cells show expression of CD68 (cytoplasmic, Golgi dot-like staining with background macrophages darkly stained), CD1a (surface), and langerin (cytoplasmic) and are negative for CD163. The mutant-specific antibody clone VE1 detects the BRAF-V600E mutation by immunohistochemistry with 2-3+ strong cytoplasmic staining. Images magnification ×400; inset magnification ×600.
The phenotype of LCH is similar to that of benign LCs, with the expression of CD68 (Golgi dot-like staining), S100 (nuclear and cytoplasmic), CD1a (surface), langerin/CD207 (cytoplasmic), and ZBTB46 (a marker of classical dendritic cells).87 They are commonly negative for monocyte-macrophage markers CD14, CD163, and OCT2.88 In contrast to reactive LCs, the neoplastic cells of LCH show more diffuse expression of cyclin D1.86 To diagnose LCH, the minimal panel of antibodies should include CD1a and langerin/CD207, with the correct pathologic pattern and correlating clinical/radiographic findings.70 In cases where BRAF-V600E is the driver mutation, this can be identified by IHC using a mutant-specific antibody clone VE1 with moderate to strong cytoplasmic staining (Figure 2).89,90
LCH can be distinguished from other histiocytic neoplasms such as ECD and RDD by morphologic and immunophenotypic features (Table 5).13,48 On morphology, ECD shows bland xanthogranulomatous inflammation and variable degrees of fibrosis,91 and RDD shows enlarged histiocytes with abundant pale-staining cytoplasm and emperipolesis. In contrast to LCH, the histiocytes of ECD and RDD express CD163 and lack expression of CD1a, langerin, and ZBTB46. ECD usually shows strong expression of factor XIIIa, and RDD is characteristically positive for S100, fascin, and OCT2.88 Mixed histiocytoses can occur with both LCH and either ECD or RDD components present within the same biopsy or involving different sites in the same patient.92 Another rare differential diagnosis to consider is indeterminate cell histiocytosis, which is likely a distinct neoplasm with the absence of langerin/CD207 expression and primarily cutaneous involvement.93 Recently, ETV3-NCOA2 translocations have been identified as a unique driver of this disease entity.94
Characteristic IHC phenotype of histiocytic disorders
| IHC Marker | LCH | RDD | ECD |
|---|---|---|---|
| CD68 | + | + | + |
| CD163 | −* | +† | + |
| Cyclin D1 | + | + | + |
| S100 | + | + | ±‡ |
| OCT2 | −* | +‖ | − |
| Factor XIIIa | − | ±§ | +‖ |
| CD1a | +‖ | − | − |
| CD207/langerin | +‖ | − | − |
| ZBTB46 | +‖ | − | − |
| IHC Marker | LCH | RDD | ECD |
|---|---|---|---|
| CD68 | + | + | + |
| CD163 | −* | +† | + |
| Cyclin D1 | + | + | + |
| S100 | + | + | ±‡ |
| OCT2 | −* | +‖ | − |
| Factor XIIIa | − | ±§ | +‖ |
| CD1a | +‖ | − | − |
| CD207/langerin | +‖ | − | − |
| ZBTB46 | +‖ | − | − |
Key differences in the phenotypes of each histiocytic disorder.
OCT2 and CD163 are expressed in a minority of LCH cases (5-10%).
CD163 is negative in a minority of RDD cases (∼10%).
S100 is expressed in ∼20-30% of ECD cases.
Factor XIIIa expression is increased in 30% of RDD cases.
The distinction between LCH and Langerhans cell sarcoma (LCS) is currently based on morphologic criteria, with the WHO classification defining LCS as having “overtly malignant cytologic features,”3,95 which may be subjective in some cases. Chromosome and molecular genetic studies may be helpful, as LCH generally has a normal karyotype and <5 somatic mutations,8 whereas malignant histiocytic neoplasms like LCS typically have frequent chromosomal gains or losses.96
Molecular analysis for somatic alterations in the MAPK-ERK and related pathways
Multiple techniques are available for molecular analysis in LCH, and a stepwise approach may be the most practical for judicious use of limited material. Given its high frequency, we recommend BRAF-V600E mutation testing in all patients (CS#16). This can be pursued by IHC or molecular methods, including quantitative polymerase chain reaction (qPCR), droplet digital PCR (ddPCR), or NGS. Allele-specific qPCR and ddPCR offer much greater sensitivity than pyrosequencing, especially given the low variant allele fractions (<5%) seen in LCH.97 The mutant-specific immunostain VE1 for BRAF-V600E has variable sensitivity and specificity in LCH tissue biopsies based on the tumor cellularity and intensity of staining (CS#17).98,99 Therefore, we suggest confirming a negative or equivocal IHC test by one of the molecular methods. BRAF-V600E mutations may also be detected by ddPCR in bronchoalveolar lavage specimens in patients with PLCH.98 For LCH specimens without BRAF-V600E, we recommend NGS for mutations and fusions in genes of the MAPK-ERK and related pathways (CS#18). In cases with insufficient tissue specimens, peripheral blood cell-free DNA testing is a reasonable alternative, bearing in mind that its correlation with tissue NGS may be higher for BRAF-V600E compared with other mutations (CS#19).100-102
Baseline evaluations
A thorough history and physical examination are essential to assess potential sites of disease and symptomatology in LCH. We recommend FDG-PET/CT imaging (skull vertex to toes) in all patients with a diagnosis or suspicion of LCH to ascertain organ involvement and target disease sites for biopsy and response assessment (CS#3). Among patients with suspected single-system PLCH, HRCT can often be diagnostic and provides comprehensive architectural details of cystic lesions seen in advanced stages of the disease (CS#6). Among patients with dyspnea or disproportionately abnormal diffusing capacity for carbon monoxide (<60% predicted), screening for pulmonary hypertension should be conducted (CS#9,10). Additional baseline laboratory and organ-specific testing should be performed as outlined in Figure 3 (CS#11-14). Among patients with otherwise unexplained peripheral blood count abnormalities, a bone marrow biopsy is warranted to rule out a concomitant hematologic neoplasm (CS#15).
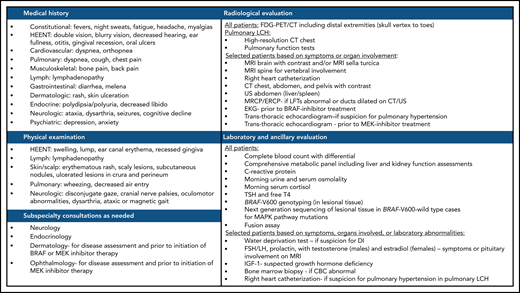
Suggested workup for a newly diagnosed or suspected LCH in adults. ACTH, adrenocorticotropic hormone; CBC, complete blood count; DI, diabetes insipidus; EKG, electrocardiogram; FSH/LH, follicle stimulating hormone/luteinizing hormone; HEENT, head, eyes, ears, nose, and throat; IGF-1, insulin-like growth factor 1; TSH, thyroid-stimulating hormone.
Suggested workup for a newly diagnosed or suspected LCH in adults. ACTH, adrenocorticotropic hormone; CBC, complete blood count; DI, diabetes insipidus; EKG, electrocardiogram; FSH/LH, follicle stimulating hormone/luteinizing hormone; HEENT, head, eyes, ears, nose, and throat; IGF-1, insulin-like growth factor 1; TSH, thyroid-stimulating hormone.
Treatment
Unifocal LCH
Unifocal LCH is often curable in adults, and local therapies may be sufficient in many cases (CS#20).103 Surgical resection may be preferred depending on the disease location but should only be attempted if not heroic or mutilating. Most skull lesions can resolve after limited curettage without the need for prostheses. If surgical resection is successful, there is no established role for adjuvant treatments. Depending on the location, intralesional corticosteroid therapy (eg, methylprednisone or triamcinolone)104 or, especially in the case of an accompanying soft tissue component, radiation therapy (eg, 10-20 Gy)105,106 may offer an excellent and durable response. If symptoms disappear quickly after diagnostic biopsy, remission can occur even without further therapy.107 Hence, in these cases, careful observation is an appropriate approach. Isolated skin nodules, lymph nodes, oral mucosal lesions, and isolated polyps of the gastrointestinal tract are usually completely resectable and require no further therapy; however, for anogenital and gingival lesions, oral methotrexate or hydroxyurea may be favored.77,108-110 Symptomatic cutaneous LCH is generally treated with topical or oral therapy. The choice of therapy depends on the severity of the disease and patient/physician preference. Topical triamcinolone can be used for disease involving small areas of the skin. Locally extensive LCH of the skin is usually less sensitive to topical or systemic steroid therapy but usually responds to treatment with hydroxyurea or low-dose methotrexate.110,111 In refractory cases, local therapy with imiquimod, 20% N-mustard, or irradiation can be effective.47,105,112,113 The treatment of unifocal LCH involving sites that are not easily amenable to local therapies (nervous system, pituitary, liver, spleen, heart, etc) is not well defined and likely warrants systemic therapies similar to multisystem disease (CS#21).
Single-system pulmonary LCH
Management of patients with single-system PLCH should be individualized and take into consideration respiratory symptoms, the degree of lung function impairment, and the radiologic extent of disease. In all cases, smoking cessation (all forms, including marijuana and vaping) is mandatory, which remains difficult to achieve in practice (CS#23). It is often the only required intervention that results in complete remission.114 Combined inhaled corticosteroid and long-acting β2-agonist therapy may provide benefit to patients with wheezing and moderate obstructive lung disease. Pneumothorax management may be particularly difficult as it may recur in half of the patients.115 Surgical pleurodesis should ideally be avoided in these young patients who may need lung transplantation in the future. Lower respiratory tract infection is a common cause of deterioration for PLCH and should be promptly treated. Seasonal influenza, pneumococcal, and COVID-19 vaccinations are recommended for all patients barring contraindications. Systemic therapy should be instituted for patients with persistent or worsening symptoms who do not respond to smoking cessation or are unable to quit smoking (CS#24). Vinblastine has limited efficacy in PLCH.116 Cladribine (2-chlorodeoxyadenosine) use was associated with improved lung function in multiple studies and is the preferred systemic therapy.117-120 Its long-term effects and tolerance are being evaluated in an uncontrolled phase 2 study (NCT01473797). The role of vasodilator therapy in severe PLCH with pulmonary hypertension is not well established and should be reserved for centers with expertise.121 Patients with advanced single-system PLCH may be candidates for lung transplantation, although the disease may relapse in the transplanted lungs (CS#25).122 The role of MAPK pathway inhibition in single-system PLCH warrants further exploration; review of existing literature showed a single case report of successful treatment using a MEK inhibitor, trametinib.123
Multifocal and multisystem LCH
Long-term remissions can be achieved with multifocal and multisystem disease, and several therapeutic principles generally apply. For patients with multisystem LCH that is asymptomatic and without critical organ involvement (eg, brain, liver, lung) or end-organ dysfunction, an initial period of careful observation and surveillance is reasonable to determine the tempo of the disease. Treatment should be initiated when symptoms ensue or if there is impending organ compromise. The optimal treatment of single-system multifocal and multisystem disease remains undefined in adult LCH because of a paucity of prospective studies. Most retrospective studies have used chemotherapy regimens adopted from the pediatric LCH experience, and some existing treatment guidelines embody this approach. However, pediatric regimens may cause greater toxicities in adults, leading to discontinuation and subsequently diminished efficacy. Moreover, the codified risk stratification system124 has not been fully validated in adults. Therefore, it is unclear whether the risk-based treatment strategy implemented in the pediatric setting can be applied to adults.
Treatment regimens that have been published in a series of ≥10 patients are listed in Table 6.110,116,120,125-130 In general, these can be categorized into 2 groups, vinca alkaloid/steroid-based and antimetabolite-based (cladribine, cytarabine) regimens. The vinca alkaloid/steroid-based regimens are similar to or adaptations of pediatric regimens. Although the response rates are generally high, between 70% and 80%, this treatment has been associated with high failure/relapse rates (40% to 80%).116,126 While 2-drug regimens are relatively well tolerated, the more complex regimens with ≥4 or more drugs have, as expected, a higher toxicity profile. The only prospective trials available are for antimetabolite-based regimens. The largest trial investigated the methotrexate + cytarabine regimen, with a response rate of 88% and a 3-year progression rate of 32%. However, almost half (48%) developed febrile neutropenia while a third (33%) developed grade 3/4 thrombocytopenia.44 The only other prospective trial used cladribine and showed a response rate of 75% with a median duration of 3 years and febrile neutropenia rate of 15%.125 Recent retrospective studies have confirmed the high response rates (79% to 90%) among patients who received cladribine as frontline or at disease relapse.120,127 Limiting cladribine treatment to 4 cycles may reduce the rate of severe cytopenias, and maximum response is attained usually within 4 cycles of treatment.120 Another nucleoside analog, clofarabine, has demonstrated efficacy in refractory pediatric LCH131 and is being evaluated in a clinical trial that includes adults (NCT02425904).
Treatment regimens for multifocal or multisystem adult LCH (series of >10 patients)
| Treatment | Dose | Type of study | n | Overall response | Median duration of response (range) | Progression rate at 3 y (%) | Comments | Reference |
|---|---|---|---|---|---|---|---|---|
| Cladribine | Cladribine 0.14 mg/kg IV d 1-5 (total of 6 cycles; 28 d/cycle) | P | 13 | 75 | 36 mo (1-65+) | ND | Newly diagnosed and relapsed/refractory settings; no patient with CNS, liver, or spleen involvement; 54% grade 3/4 neutropenia; 15% febrile neutropenia; no grade 3/4 anemia or thrombocytopenia | Saven 1999125 |
| Cladribine 5 mg/m2 IV d 1-5 (total of 6 cycles; 28 d/cycle) | R | 22 | 41* | ND | ND | Newly diagnosed and relapsed/refractory settings; all with unifocal or multifocal bone lesions ± other organ involvement; grade 3/4 cytopenias, 22% | Cantu 2012126 | |
| Cladribine 0.14 mg/kg IV d 1-5 (3-6 cycles; 28 d/cycle) | R | 22 | 88 | ND | 35 | Relapsed/refractory setting; 56% nonpituitary CNS, 43% lung, and 13% nodal involvement; 46% CR and 46% PR among those with CNS disease | Néel 2020127 | |
| Cladribine 5 mg/m2 or 0.14 mg/kg IV d 1-5 or (1-9 cycles; 28 d/cycle) | R | 38 | 79 | NR (5y, 70%) | Newly diagnosed and relapsed/refractory settings; 60% lung, 30% lymph nodes, 27% pituitary; 26% CR, 53% PR; responses seen in all disease sites; PFS similar between CR and PR patients; trend toward longer PFS in non-BRAF V600E | Goyal 2021120 | ||
| CEVP | Cyclophosphamide 750 mg/m2 IV day 1; etoposide 100 mg/m2 IV d 1-3; vindesine 4 mg IV d 1; prednisone 100 mg PO d 1-5 (median 5 cycles; 21 d/cycle) | R | 31 | 70 | ND | 20 | Newly diagnosed and relapsed/refractory settings; 48% lung, 19% liver, and 7% spleen involvement; organ-specific responses not reported; any neutropenia, 48%; any thrombocytopenia, 13%; any hepatotoxicity, 23% | Duan 2016128 |
| Cytarabine | 100 mg/m2 IV d 1-5 (6 cycles; 28 d/cycle) | R | 24 | 79* | ND | ND | Newly diagnosed and relapsed/refractory settings; all with unifocal or multifocal bone lesions ± other organ involvement; grade 3/4 cytopenias, 20% | Cantu 2012126 |
| Hydroxyurea | Hydroxyurea 500 mg PO bid | R | 10 | 60 | ND | 40 | Restricted to skin, oral, bone, or lymph node limited disease | Zinn 2016110 |
| MACOP-B | Cyclophosphamide 350 mg/m2 IV + doxorubicin 50 mg/m2 IV d 1, 15, 29, 43, and 71; methotrexate 400 mg/m2 IV d 8, 36, and 64 with leucovorin rescue vincristine 1.4 mg/m2 IV d 8, 22, 36, 50, and 64; bleomycin 10 mg/m2 IV d 22, 50, and 78; prednisone 40 mg/m2 PO d 1-84 | R | 11 | 100 | 47+ months (5-228+) | 30 | Newly diagnosed and relapsed/refractory settings; 36% lung and 9% spleen involvement; organ-specific responses not reported; grade 3 neutropenia, 36%; grade 3 hepatotoxicity, 9%; no febrile neutropenia | Derenzini 2015129 |
| Methotrexate + cytarabine (MC) | Methotrexate 1 g/m2 IV over 24 h day 1 cytarabine 100 mg/m2 IV over 24 h d 1-5 (6 cycles; 35 d/cycle) | P | 83 | 88 | ND | 32 | Newly diagnosed setting; 68% lung, 28% liver, 13% spleen, and 4% nonpituitary CNS involvement; organ-specific responses not reported; 94% grade 3/4 neutropenia; 33% grade 3/4 thrombocytopenia; 48% febrile neutropenia; no treatment related death | Cao 202044 |
| Vinblastine + prednisone (VbP) | Vinblastine 6 mg/m2 (10 mg maximum) IV weekly × 6, followed by maintenance phase dosing every 3 wk × 6-12 mo; prednisone 40 mg/m2 PO daily × 4 wk, then taper, followed by 40 mg/m2 PO maintenance phase dosing d 1-5 every 3 wk × 6-12 mo | R | 19 | 16* | ND | ND | Newly diagnosed and relapsed/refractory settings; all with unifocal or multifocal bone lesions ± other organ involvement; grade 3/4 neuropathy, 75% | Cantu 2012126 |
| R | 35 | 71 | ND | 35 | Newly diagnosed and relapsed/refractory settings; 30% lung, 15% liver, 7% nonpituitary CNS, and 7% nodal involvement; organ-specific responses not reported; grade 3/4 neutropenia, 17%; grade 2 neuropathy, 26% | Tazi 2017116 | ||
| VbPMM | Vinblastine 6 mg/m2 (6 mg maximum) IV d 1; prednisolone 2 mg/kg (60 mg maximum) PO d 1-5; methotrexate 20 mg/m2 PO d 15; 6-mercaptopurine 1.5 mg/kg PO d 1-28 (9 cycles; 28 d/cycle) | R | 13 | 85 | ND | 45 | Newly diagnosed and relapsed/refractory settings; 21% lung, 21% nodal, and 7% nonpituitary CNS involvement; organ-specific responses not reported; grade 3/4 neutropenia, 21%; febrile neutropenia, 7%; grade 3 hepatotoxicity, 7%; grade 5 bleeding, 7% | Morimoto 2013130 |
| Vindesine + prednisone (VP) | Vindesine 4 mg IV d 1; prednisone 100 mg PO d 1-5 (median 5 cycles; 21 d/cycle) | R | 14 | 64 | ND | 40 | Newly diagnosed and relapsed/refractory settings; 50% lung, 7% liver, and 14% spleen involvement; organ-specific responses not reported; any neutropenia, 7%; no thrombocytopenia; any hepatotoxicity, 7% | Duan 2016128 |
| Treatment | Dose | Type of study | n | Overall response | Median duration of response (range) | Progression rate at 3 y (%) | Comments | Reference |
|---|---|---|---|---|---|---|---|---|
| Cladribine | Cladribine 0.14 mg/kg IV d 1-5 (total of 6 cycles; 28 d/cycle) | P | 13 | 75 | 36 mo (1-65+) | ND | Newly diagnosed and relapsed/refractory settings; no patient with CNS, liver, or spleen involvement; 54% grade 3/4 neutropenia; 15% febrile neutropenia; no grade 3/4 anemia or thrombocytopenia | Saven 1999125 |
| Cladribine 5 mg/m2 IV d 1-5 (total of 6 cycles; 28 d/cycle) | R | 22 | 41* | ND | ND | Newly diagnosed and relapsed/refractory settings; all with unifocal or multifocal bone lesions ± other organ involvement; grade 3/4 cytopenias, 22% | Cantu 2012126 | |
| Cladribine 0.14 mg/kg IV d 1-5 (3-6 cycles; 28 d/cycle) | R | 22 | 88 | ND | 35 | Relapsed/refractory setting; 56% nonpituitary CNS, 43% lung, and 13% nodal involvement; 46% CR and 46% PR among those with CNS disease | Néel 2020127 | |
| Cladribine 5 mg/m2 or 0.14 mg/kg IV d 1-5 or (1-9 cycles; 28 d/cycle) | R | 38 | 79 | NR (5y, 70%) | Newly diagnosed and relapsed/refractory settings; 60% lung, 30% lymph nodes, 27% pituitary; 26% CR, 53% PR; responses seen in all disease sites; PFS similar between CR and PR patients; trend toward longer PFS in non-BRAF V600E | Goyal 2021120 | ||
| CEVP | Cyclophosphamide 750 mg/m2 IV day 1; etoposide 100 mg/m2 IV d 1-3; vindesine 4 mg IV d 1; prednisone 100 mg PO d 1-5 (median 5 cycles; 21 d/cycle) | R | 31 | 70 | ND | 20 | Newly diagnosed and relapsed/refractory settings; 48% lung, 19% liver, and 7% spleen involvement; organ-specific responses not reported; any neutropenia, 48%; any thrombocytopenia, 13%; any hepatotoxicity, 23% | Duan 2016128 |
| Cytarabine | 100 mg/m2 IV d 1-5 (6 cycles; 28 d/cycle) | R | 24 | 79* | ND | ND | Newly diagnosed and relapsed/refractory settings; all with unifocal or multifocal bone lesions ± other organ involvement; grade 3/4 cytopenias, 20% | Cantu 2012126 |
| Hydroxyurea | Hydroxyurea 500 mg PO bid | R | 10 | 60 | ND | 40 | Restricted to skin, oral, bone, or lymph node limited disease | Zinn 2016110 |
| MACOP-B | Cyclophosphamide 350 mg/m2 IV + doxorubicin 50 mg/m2 IV d 1, 15, 29, 43, and 71; methotrexate 400 mg/m2 IV d 8, 36, and 64 with leucovorin rescue vincristine 1.4 mg/m2 IV d 8, 22, 36, 50, and 64; bleomycin 10 mg/m2 IV d 22, 50, and 78; prednisone 40 mg/m2 PO d 1-84 | R | 11 | 100 | 47+ months (5-228+) | 30 | Newly diagnosed and relapsed/refractory settings; 36% lung and 9% spleen involvement; organ-specific responses not reported; grade 3 neutropenia, 36%; grade 3 hepatotoxicity, 9%; no febrile neutropenia | Derenzini 2015129 |
| Methotrexate + cytarabine (MC) | Methotrexate 1 g/m2 IV over 24 h day 1 cytarabine 100 mg/m2 IV over 24 h d 1-5 (6 cycles; 35 d/cycle) | P | 83 | 88 | ND | 32 | Newly diagnosed setting; 68% lung, 28% liver, 13% spleen, and 4% nonpituitary CNS involvement; organ-specific responses not reported; 94% grade 3/4 neutropenia; 33% grade 3/4 thrombocytopenia; 48% febrile neutropenia; no treatment related death | Cao 202044 |
| Vinblastine + prednisone (VbP) | Vinblastine 6 mg/m2 (10 mg maximum) IV weekly × 6, followed by maintenance phase dosing every 3 wk × 6-12 mo; prednisone 40 mg/m2 PO daily × 4 wk, then taper, followed by 40 mg/m2 PO maintenance phase dosing d 1-5 every 3 wk × 6-12 mo | R | 19 | 16* | ND | ND | Newly diagnosed and relapsed/refractory settings; all with unifocal or multifocal bone lesions ± other organ involvement; grade 3/4 neuropathy, 75% | Cantu 2012126 |
| R | 35 | 71 | ND | 35 | Newly diagnosed and relapsed/refractory settings; 30% lung, 15% liver, 7% nonpituitary CNS, and 7% nodal involvement; organ-specific responses not reported; grade 3/4 neutropenia, 17%; grade 2 neuropathy, 26% | Tazi 2017116 | ||
| VbPMM | Vinblastine 6 mg/m2 (6 mg maximum) IV d 1; prednisolone 2 mg/kg (60 mg maximum) PO d 1-5; methotrexate 20 mg/m2 PO d 15; 6-mercaptopurine 1.5 mg/kg PO d 1-28 (9 cycles; 28 d/cycle) | R | 13 | 85 | ND | 45 | Newly diagnosed and relapsed/refractory settings; 21% lung, 21% nodal, and 7% nonpituitary CNS involvement; organ-specific responses not reported; grade 3/4 neutropenia, 21%; febrile neutropenia, 7%; grade 3 hepatotoxicity, 7%; grade 5 bleeding, 7% | Morimoto 2013130 |
| Vindesine + prednisone (VP) | Vindesine 4 mg IV d 1; prednisone 100 mg PO d 1-5 (median 5 cycles; 21 d/cycle) | R | 14 | 64 | ND | 40 | Newly diagnosed and relapsed/refractory settings; 50% lung, 7% liver, and 14% spleen involvement; organ-specific responses not reported; any neutropenia, 7%; no thrombocytopenia; any hepatotoxicity, 7% | Duan 2016128 |
CR, complete remission; PR, partial remission; ND, not defined; P, prospective; R, retrospective.
Composite endpoint including patients who did not respond or progressed at 1 y.
Other systemic treatments, including bisphosphonates for bone-predominant disease,132 immunomodulatory imide drugs (IMiDs),133-135 hydroxyurea, oral low-dose methotrexate, hematopoietic stem cell transplant (HSCT), and BRAF and MEK inhibitors, have been reported in smaller series or case reports. In a combined adult and pediatric series of patients with skeletal disease, the use of bisphosphonate therapy (alendronate, pamidronate, or zoledronate) was associated with durable resolution of bone pains as well as radiographic ossification and normalization of bone lesions in the majority of the cases (92%). About half of the patients received only 1 infusion of zoledronate, and the rest were treated over months to a few years.132 Of the IMiDs, both thalidomide and lenalidomide have been reported to elicit responses, with some lasting over a year. However, maintenance therapy was required, and relapses occurred almost uniformly after discontinuation of therapy.133,134 Multifocal skin disease is usually responsive to hydroxyurea, IMiDs, oral low-dose methotrexate therapy with or without prednisone/6-mercaptopurine.110,111,133,136 Patients with asymptomatic lymph node-only disease can be monitored closely without treatment and may result in spontaneous remissions, especially in the case of cervical lymphadenopathy.109 There are 6 published cases of adults with refractory LCH (age range 21-35 years) who underwent HSCT, with most cases achieving sustained remissions.136-141 Data on the efficacy of BRAF and MEK inhibitors in frontline and relapsed setting are emerging with several published cases. The majority had BRAF-V600E mutation and received vemurafenib, although there are also reports using cobimetinib, trametinib, and dabrafenib + trametinib.9,10,136,142-146 Responses were near-universal regardless of the site of involvement, and most had a sustained complete remission as long as treatment continued.
Suggested treatment approach
For unifocal disease, local therapy such as surgical excision may be curative if it can be safely undertaken. Unifocal disease not amenable to local therapies (pituitary, CNS, heart) should be treated similar to multisystem disease with systemic therapies (CS#21). Radiation therapy is an effective option for symptomatic bone-only disease with a limited number of lesions (<3). Other treatment options for multifocal cutaneous or bone disease include immunosuppressive agents like low-dose methotrexate, hydroxyurea, 6-mercaptopurine, or IMiDs. Bisphosphonates are an option for bone-only disease without the risk of structural compromise or instability (CS#26-27). For disease recurrence not amenable to local therapies or unresponsive to immunosuppressive agents, systemic chemotherapy is recommended (Table 6; CS#28). The treatment of single-system PLCH often requires collaboration between pulmonologists and oncologists. For multifocal and multisystem disease, we propose the treatment approach using the algorithm provided in Figure 4. Among systemic treatment options for LCH, chemotherapy using either cladribine or cytarabine is preferred because of relatively high overall response rates and the potential for long-term remissions with limited cycles of treatment (CS#28). Vinblastine + prednisone is a reasonable alternative, although it is not the preferred choice of therapy in adults given the high risk of relapse and potential for peripheral neuropathy. Data on BRAF and MEK inhibitors are still evolving, but advantages include near-universal and quick responses, making them a preferred treatment for patients who require a rapid reversal of organ compromise. Similar to the experience in ECD, discontinuation of these agents will likely result in disease relapse, necessitating prolonged therapy.147 However, carefully selected patients may be good candidates for low-dose chronic administration or treatment breaks. Because most patients will require a dose reduction and lower doses of BRAF and MEK inhibitors are quite effective, it is reasonable to start at half of the dose currently used to treat patients with melanoma. With the advent of targeted inhibitors, the role of HSCT in adults with refractory LCH is unclear.
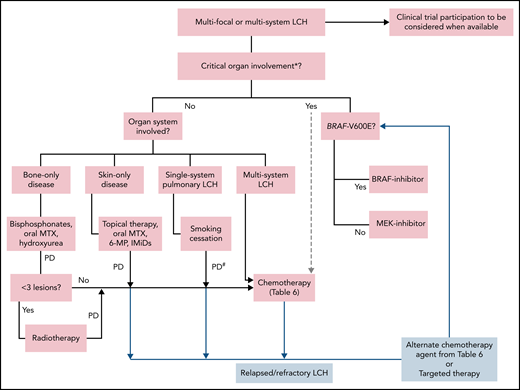
Treatment algorithm for adults with multifocal or multisystem LCH. Systemic therapy is indicated for patients with single-system unifocal disease involving critical organs or specific sites (nervous system, liver, spleen, etc). *Brain (esp. neurodegenerative LCH), liver (esp. sclerosing cholangitis). Liver transplant consult for sclerosing cholangitis. #Systemic therapy may be indicated in patients with symptomatic disease and unable to quit smoking. Lung transplantation referral should be undertaken if not eligible for or refractory to systemic treatments. 6-MP, 6-mercaptopurine; IMiDs, immunomodulators (thalidomide, lenalidomide); MTX, methotrexate; PD, progressive disease.
Treatment algorithm for adults with multifocal or multisystem LCH. Systemic therapy is indicated for patients with single-system unifocal disease involving critical organs or specific sites (nervous system, liver, spleen, etc). *Brain (esp. neurodegenerative LCH), liver (esp. sclerosing cholangitis). Liver transplant consult for sclerosing cholangitis. #Systemic therapy may be indicated in patients with symptomatic disease and unable to quit smoking. Lung transplantation referral should be undertaken if not eligible for or refractory to systemic treatments. 6-MP, 6-mercaptopurine; IMiDs, immunomodulators (thalidomide, lenalidomide); MTX, methotrexate; PD, progressive disease.
For patients with CNS disease, cladribine, higher doses of cytarabine, IV methotrexate-based regimens, or kinase inhibitors are preferred (CS#29,30). The treatment data on adults with neurodegenerative disease are lacking, but BRAF/MEK–inhibitor- or cytarabine-based chemotherapy may be preferred based on limited pediatric experience.148,149 For LCH-associated sclerosing cholangitis, the optimum therapy is unknown. Due to poor outcomes with chemotherapy, targeted agents are preferable. Prolonged remissions following liver transplant have been reported50,150-152; therefore, early transplant referral should be undertaken in otherwise fit patients. In a study of mostly pediatric patients with LCH, disease relapse in the transplanted liver was seen in 8% of cases.152 For disease that is relapsed or refractory following chemotherapy, it is reasonable to consider an alternate chemotherapy agent or a kinase inhibitor based on patient-specific factors and drug availability (CS#30).
Response assessment and monitoring
Response assessment and surveillance strategies in LCH vary based on multiple factors: organ involvement at diagnosis, type of treatment used, and patient preference (CS#31). For patients with abnormal FDG-PET/CT findings at diagnosis, the first response assessment should occur in 2 to 3 months of initiation of therapy and every 3 to 6 months thereafter (CS#32). In select cases, organ-specific imaging (CT, HRCT, ultrasound, or magnetic resonance imaging) may be necessary to assess for a response. Additional testing may be necessary based on clinical manifestations and organs involved, with specific tailoring for single-system PLCH (Table 8). There are no established response criteria for adults with LCH, and pediatric criteria set forth by the International LCH Study Group153 may not be applicable in adults. Recent adult histiocytosis clinical trials and retrospective studies have used clinical and radiographic criteria that are preferred in adult LCH (Table 7).9,10,154 Any degree of improvement in clinical or radiographic findings is considered a response. It may take several months to attain the best response, especially with oral therapies. Once the best response is achieved and disease has stabilized, the frequency of imaging studies can be individualized. As endocrinopathies involving the pituitary or hypothalamus may not resolve with LCH-specific therapy, it is necessary to monitor the hormone levels and continue replacement, preferably in collaboration with an endocrinologist.
Radiographic response criteria for adults with LCH
| Response category* | Radiographic response criteria (FDG PET) | Radiographic response criteria (CT or MRI) |
|---|---|---|
| Complete response | Normalization of lesions with FDG uptake equal to surrounding background tissue | Complete anatomic resolution of lesions or resolution of abnormal imaging features (enhancement, diffusion restriction, etc)† |
| Partial response | Reduction from baseline SUV of lesions, but persistent uptake greater than surrounding background tissue | Reduction, but not complete resolution of lesions/abnormal imaging features |
| Progressive disease | Increased SUV value of lesions as compared with before or appearance of new FDG avid lesions | Worsening of abnormal imaging features or growth of existing lesions or appearance of new lesions |
| Stable disease | Does not meet other criteria | Does not meet other criteria |
| Response category* | Radiographic response criteria (FDG PET) | Radiographic response criteria (CT or MRI) |
|---|---|---|
| Complete response | Normalization of lesions with FDG uptake equal to surrounding background tissue | Complete anatomic resolution of lesions or resolution of abnormal imaging features (enhancement, diffusion restriction, etc)† |
| Partial response | Reduction from baseline SUV of lesions, but persistent uptake greater than surrounding background tissue | Reduction, but not complete resolution of lesions/abnormal imaging features |
| Progressive disease | Increased SUV value of lesions as compared with before or appearance of new FDG avid lesions | Worsening of abnormal imaging features or growth of existing lesions or appearance of new lesions |
| Stable disease | Does not meet other criteria | Does not meet other criteria |
SUV, standardized uptake value.
For LCH without radiographic abnormalities (e.g., skin, gastrointestinal tract), clinical criteria (physical exam, endoscopy) should be used to assess response.
Patients with endocrinopathies from pituitary involvement may require ongoing hormone replacement despite attaining a radiographic complete response. Patients with single- system pulmonary LCH and abnormal PFTs at baseline should undergo follow-up testing to assess response.
Response assessment and disease surveillance recommendations for LCH in adults
| Type of LCH | First response assessment | Subsequent assessments | Comments |
|---|---|---|---|
| Skin only | Skin exam at each visit | Skin exam at each visit | |
| Bone only | FDG-PET/CT in 2-3 mo | FDG-PET/CT every 3-6 mo* | |
| Pituitary and hypothalamus only | Endocrine evaluation in 3 mo | Endocrine evaluation annually | MRI of pituitary/hypothalamus if initially involved |
| Other unifocal LCH | FDG-PET/CT or CT/MRI in 2-3 mo | FDG-PET/CT or CT/MRI every 3-6 mo* | |
| Single-system PLCH | PFTs and HRCT chest in 3-6 mo† | PFTs and HRCT chest 6-12 mo† | TTE and right-heart catheterization may be needed if suspicion of pulmonary hypertension |
| Multisystem LCH | FDG-PET/CT in 2-3 mo | FDG-PET/CT every 3-6 mo* | Organ-specific imaging (CT or MRI) may be needed depending on organ involvement |
| Type of LCH | First response assessment | Subsequent assessments | Comments |
|---|---|---|---|
| Skin only | Skin exam at each visit | Skin exam at each visit | |
| Bone only | FDG-PET/CT in 2-3 mo | FDG-PET/CT every 3-6 mo* | |
| Pituitary and hypothalamus only | Endocrine evaluation in 3 mo | Endocrine evaluation annually | MRI of pituitary/hypothalamus if initially involved |
| Other unifocal LCH | FDG-PET/CT or CT/MRI in 2-3 mo | FDG-PET/CT or CT/MRI every 3-6 mo* | |
| Single-system PLCH | PFTs and HRCT chest in 3-6 mo† | PFTs and HRCT chest 6-12 mo† | TTE and right-heart catheterization may be needed if suspicion of pulmonary hypertension |
| Multisystem LCH | FDG-PET/CT in 2-3 mo | FDG-PET/CT every 3-6 mo* | Organ-specific imaging (CT or MRI) may be needed depending on organ involvement |
PFT, pulmonary function tests; TTE, transthoracic echocardiogram.
Once the best response is achieved and disease stabilized, the frequency of testing and imaging studies can be individualized to longer intervals.
Chest radiography can be performed instead of HRCT for follow-up in cases that are mild and do not require treatment. Serial lung HRCT is recommended in patients who exhibit changes in their clinical, functional, or chest radiography findings during follow-up.
Chronic pain, fatigue, and quality of life
In the collective experience of the authors, patients with LCH frequently endorse disabling chronic musculoskeletal pain, fatigue, and mental fogging that diminish their quality of life. In some instances, previously healthy adults who develop a discrete single system lesion of LCH find that they never return to full functional status. In the experience of the authors, the site of a bone lesion remains an enduring focus of pain in up to half of patients despite the resolution of metabolic activity on PET/CT imaging. In many cases, the constellation of symptoms satisfies the criteria for the diagnosis of chronic fatigue syndrome155-157 or fibromyalgia.158 Such patients may benefit from referral to specialized services, if available.
In new patients, caution should be exercised in promising complete resolution of symptoms, and timely preparation for lifelong symptom management is warranted. Patients with persistent pain will typically achieve unsatisfactory responses to most available analgesic drugs. Some patients may notice a relief of fatigue with methylphenidate, which appears to be safe for use in the long term. Optimum management of concomitant depression and anxiety is also recommended and may improve quality of life, fatigue, and the burden of chronic pain. The experience of the authors is that, in many instances, unmanageable symptoms of patients are incorrectly viewed as a result of a maladaptive psychological state, while the reverse is, in reality, more likely. Physicians specializing in histiocytosis should acknowledge and validate the patient’s experience of these problems as “real” (ie, in some way related to disease) even if they are difficult to fully palliate. This is often met with lasting appreciation even if therapeutic options are limited. Lack of recognition of the severity of chronic ill health is a problem for many patients who outwardly look well; a reminder that LCH is classified as a hematologic neoplasm is often useful when empathy or services are not forthcoming. Early referral for psychological support and counseling for coping with chronic pain is recommended and may be the most beneficial strategy for many patients.
Prognosis
The prognosis of LCH depends on the extent of organ involvement and the response to treatment. Unifocal LCH has an excellent prognosis, with 5-year overall survival of >90%. In a series of 44 unifocal LCH cases, 20% had disease recurrence at another site but were able to be successfully treated.103 The natural history of single-system PLCH is variable and difficult to predict; ∼50% of patients experience a decrease of pulmonary function at 5 years of follow-up, and some will develop obstructive lung disease.159 The 5-year overall survival of single-system PLCH was reported to be around 70% to 80% in earlier studies,33,41 and seems more favorable in the current era.13,160 Approximately half of the deaths in PLCH occur due to respiratory failure,33 and follow-up testing is essential to identify patients who may develop progressive disease. The prognosis of multisystem/disseminated LCH is generally considered good with ∼90% 5-year overall survival rates.4,41 A recent phase 2 trial of methotrexate and cytarabine suggested that liver and spleen involvement may be associated with poorer prognosis in adults, although none of the patients received BRAF or MEK inhibitors.44 The prognosis of rare phenotypes such as sclerosing cholangitis and neurodegenerative LCH is guarded but may improve in the era of targeted therapies. As the LCH-specific survival of adult patients improves with novel therapeutics, it is critical to evaluate and monitor them for long-term effects of therapies and disease, including new-onset morbidities and second cancers that may result in premature mortality.
Acknowledgment
This work was supported by the Walter B. Frommeyer, Jr., Fellowship Award in Investigative Medicine, University of Alabama at Birmingham (G.G.).
Authorship
Contribution: All authors participated in outlining the manuscript and providing expert recommendation grading; G.G. wrote the manuscript draft with input from R.S.G., K.L.R., J.L.P., R.V., J.V.L., M.L.H., X.-X.C., P.M., G.K., J.H., M.C., and K.L.M., E.L.D., and M.G., ; J.R.Y., A.T., and C.W.C. provided the clinical and radiographic images; and all authors then contributed to the editing of the manuscript.
Conflict of interest disclosures: M.L.H. is a member of the external advisory board at Novartis and Sobi Corporation; P.M. received fees for lectures/advisory boards and research grants from Amgen and fees for lectures/advisory boards from Glaxo, Lilly, UCB, Elpen, and Vianex. G.K. received honorarium and research grants from IPSEN, Novartis, Pfizer, Novo, and Shire. K.L.M. is a member of the medical advisory board for SOBI Corporation. E.L.D. received editorial support from Pfizer, Inc, and paid advisory board membership with Day One Biopharmaceuticals. All remaining authors declare no competing financial interests.
Correspondence: Eli L. Diamond, Department of Neurology, Memorial Sloan-Kettering Cancer Center, 160 East 53rd St, 2nd Floor Neurology, New York, NY 10022; e-mail: diamone1@mskcc.org; and Michael Girschikofsky, Internal Medicine I (Hemostasis, Hematology and Stem Cell Transplantation and Medical Oncology), Ordensklinikum Linz Elisabethinen, Fadingerstr 1, 4020 Linz, Austria; e-mail: michael.girschikofsky@ordensklinikum.at.
The online version of this article contains a data supplement.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal