

Key Points
Crystal structures of GPVI bound to collagen peptides reveal its primary collagen-binding site across the D1 domain β-sheet.
GPVI binds sites in collagen formed by two of the three triple-helix chains and canonical OGPOGP sequence motifs.

Abstract
Glycoprotein VI (GPVI) mediates collagen-induced platelet activation after vascular damage and is an important contributor to the onset of thrombosis, heart attack, and stroke. Animal models of thrombosis have identified GPVI as a promising target for antithrombotic therapy. Although for many years the crystal structure of GPVI has been known, the essential details of its interaction with collagen have remained elusive. Here, we present crystal structures of the GPVI ectodomain bound to triple-helical collagen peptides, which reveal a collagen-binding site across the β-sheet of the D1 domain. Mutagenesis and binding studies confirm the observed binding site and identify Trp76, Arg38, and Glu40 as essential residues for binding to fibrillar collagens and collagen-related peptides (CRPs). GPVI binds a site on collagen comprising two collagen chains with the core formed by the sequence motif OGPOGP. Potent GPVI-binding peptides from Toolkit-III all contain OGPOGP; weaker binding peptides frequently contain a partial motif varying at either terminus. Alanine-scanning of peptide III-30 also identified two AGPOGP motifs that contribute to GPVI binding, but steric hindrance between GPVI molecules restricts the maximum binding capacity. We further show that no cooperative interactions could occur between two GPVI monomers binding to a stretch of (GPO)5 and that binding of ≥2 GPVI molecules to a fibril-embedded helix requires non-overlapping OGPOGP motifs. Our structure confirms the previously suggested similarity in collagen binding between GPVI and leukocyte-associated immunoglobulin-like receptor 1 (LAIR-1) but also indicates significant differences that may be exploited for the development of receptor-specific therapeutics.
Introduction
Collagen exposed at sites of vascular injury contributes to thrombus formation. In response to vessel damage, platelet receptors GPIbα and integrin α2β1 support firm platelet adhesion to collagens of the subendothelial extracellular matrix (ECM): GPIbα associates with collagen-bound von Willebrand factor (VWF) whereas integrin α2β1 binds directly to exposed collagen.1,2 Subsequent interaction between collagen and the glycoprotein VI–Fc-receptor γ-chain complex3-5 triggers phosphorylation of FcRγ, ultimately resulting in full platelet activation.6-9 GPVI itself is present in both monomeric and dimeric forms in the platelet membrane, and activation by several ligands shifts the population toward the dimeric species, detected by dimer-specific antibodies.10,11 Deficiency or inhibition of GPVI results in decreased thrombus deposition and platelet aggregation,12 emphasizing its role in platelet activation.
Fibrillar collagens provide strength to the arterial vasculature, while the role of exposed collagens in platelet adhesion and activation reflects its hemostatic function.13 The triple-helical structure of collagen requires the repeating Gly-X-Y sequence, where, most frequently, X is proline (P), and Y is the post-translationally modified 4-hydroxyproline (O). Toolkit-II and -III contain homotrimeric peptides covering the full sequences of the collagen II and III triple-helical regions, respectively, and were used to identify collagenous binding motifs for several proteins,14 including integrin α2β1,15 ECM protein SPARC,16,17 discoidin domain receptor 2,18 the VWF A3 domain,19,20 and the osteoclast-associated receptor (OSCAR).21 Binding of GPVI to Toolkit-III peptides10,22 is notably more scattered than observed for other receptors.14 In addition to several peptides that support only modest GPVI binding, III-30, III-01, and III-40 exhibit high affinity for GPVI.10 High imino acid content is not restricted to these peptides; nevertheless, the existence of sequential GPO/OGP triplets appears a key determinant for GPVI-mediated platelet activation.22 These conclusions are further supported by the increasing GPVI binding capacity of model peptides containing consecutive GPO repeats, ranging from (GPO)2 to (GPO)10, the latter being the collagenous domain of collagen-related peptide (CRP).23,24 However, these studies could not unambiguously identify a GPVI binding motif.10,22
GP6 is located in chromosome 19 in the leukocyte receptor complex5 alongside many other immune receptor genes, including the structurally related but functionally separate collagen-binding receptors, OSCAR25 and leukocyte-associated immunoglobulin-like receptors 1 and 2 (LAIR-1 and -2).26 Activatory receptors GPVI and OSCAR share the same extracellular domain architecture, having two C2-type Ig-like domains (D1 and D2). Collagen binds both domains of OSCAR,21 with D2 having higher affinity, whereas binding to GPVI was reported to be primarily mediated by the hinge region of its D1 domain.27-30 LAIRs are immune-regulating receptors and have a single Ig-like domain (D1) that contains a collagen-binding site across the β-sheet formed by strands FCC’.31 Some LAIR–1-binding Toolkit peptides also bind either GPVI or OSCAR, but not both, suggesting discrete collagen-binding mechanisms for each receptor.10,22,25,32
Here, we present crystal structures of GPVI in complex with collagen-like peptides that provide the structural basis for collagen binding to the canonical OGPOGP sequence of collagen and account for the propensity of GPVI to bind diverse sites in collagen.
Methods
Expression and purification of GPVI D1D2 wild type (WT) and mutants
For crystallization, GPVI loop truncation mutants were made in a construct encoding the D1D2 domains (Uniprot-ID Q9HCN6-3; Gln21-Ser206), using a QuikChange Site-Directed Mutagenesis Kit and the primers listed in supplemental Table 1, available on the Blood Web site. GPVI was expressed with N-terminal cystatin-S signal peptide (Uniprot-ID P01036; Met1-Ala20) followed by TEV-cleavable His6-StrepII3-tag in HEK293-EBNA1-S cells (U-Protein Express). Proteins were purified on a Strep-Tactin column (GE Healthcare) and eluted with buffer containing 10 mM HEPES; pH, 7.5; 150 mM NaCl; and 5 mM d-desthiobiotin. Purification tags were removed by overnight incubation with 1:30 mol: mol of His6-TEV-protease:GPVI in the same buffer. Uncleaved protein and TEV protease were removed by immobilized metal affinity chromatography. Proteins were purified further by size-exclusion chromatography on a Superdex75 column (GE Healthcare) equilibrated in 10 mM HEPES; pH, 7.5; 150 mM NaCl; and concentrated to 10 mg/mL.
For binding studies, the WT GPVI fragment described above and point-mutants generated by QuikChange site-directed mutagenesis were transiently expressed with N-terminal cystatin-S signal peptide and C-terminal TEV-cleavable immunoglobulin Fc-tag (Uniprot ID P01857; Glu99-Lys330) in HEK293-EBNA1 cells (U-Protein Express). GPVI-Fc dimers were purified by Protein A affinity chromatography (GE Healthcare) eluted with 0.1 M glycine; pH, 3.0; fractions were collected into 1/10 volume of neutralization buffer (1 M Tris-HCl pH 9.0). Protein purity was determined as >95% by Sodium-dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were stored in neutralized elution buffer at 277 K or, for long-term storage, at 193 K after flash-freezing in liquid nitrogen.
Peptide synthesis and melting point determination
Peptides were synthesized in an Applied Biosystems Pioneer solid-phase synthesizer, using Fmoc chemistry as previously described,15,22 or were from CambCol Laboratories (Ely, United Kingdom). Peptides were purified by high-performance liquid chromatography and verified using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry. To verify the triple-helical authenticity of the peptides in solution, Tm was determined by polarimetry15 (Table 1). Tm represents the temperature at which half of the peptide has unfolded from triple-helical to random coil conformation, and high Tm indicates an accurate collagen-like structure.
Primary sequences of collagen model peptides
| Peptide | Sequence | Tm (°C) |
|---|---|---|
| (GPO)5 | (GPO)5-NH2 | 28.2 |
| (GPO)3 | (GPP)2(GPO)3(GPP)2-NH2 | 41.1 |
| CRP | GCO(GPO)10GCOG-NH2 | 82.3 |
| III30 | GPC(GPP)5GAOGLRGGAGPOGPEGGKGAAGPOGPO(GPP)5GPC-NH2 | 47.4 |
| III30-A3 | GPC(GPP)5GAAGLRGGAGPOGPEGGKGAAGPOGPO(GPP)5GPC-NH2 | 39.0 |
| III30-A5 | GPC(GPP)5GAOGARGGAGPOGPEGGKGAAGPOGPO(GPP)5GPC-NH2 | 46.9 |
| III30-A6 | GPC(GPP)5GAOGLAGGAGPOGPEGGKGAAGPOGPO(GPP)5GPC-NH2 | 44.9* |
| III30-A8 | GPC(GPP)5GAOGLRGAAGPOGPEGGKGAAGPOGPO(GPP)5GPC-NH2 | 44.8* |
| III30-A11 | GPC(GPP)5GAOGLRGGAGAOGPEGGKGAAGPOGPO(GPP)5GPC-NH2 | 41.3 |
| III30-A12 | GPC(GPP)5GAOGLRGGAGPAGPEGGKGAAGPOGPO(GPP)5GPC-NH2 | 42.2 |
| III30-A14 | GPC(GPP)5GAOGLRGGAGPOGAEGGKGAAGPOGPO(GPP)5GPC-NH2 | 43.3 |
| III30-A15 | GPC(GPP)5GAOGLRGGAGPOGPAGGKGAAGPOGPO(GPP)5GPC-NH2 | 47.4 |
| III30-A17 | GPC(GPP)5GAOGLRGGAGPOGPEGAKGAAGPOGPO(GPP)5GPC-NH2 | 44.8* |
| III30-A18 | GPC(GPP)5GAOGLRGGAGPOGPEGGAGAAGPOGPO(GPP)5GPC-NH2 | 45.5 |
| III30-A23 | GPC(GPP)5GAOGLRGGAGPOGPEGGKGAAGAOGPO(GPP)5GPC-NH2 | 45.0 |
| III30-A24 | GPC(GPP)5GAOGLRGGAGPOGPEGGKGAAGPAGPO(GPP)5GPC-NH2 | 44.4 |
| III30-A26 | GPC(GPP)5GAOGLRGGAGPOGPEGGKGAAGPOGAO(GPP)5GPC-NH2 | 47.2 |
| III30-A27 | GPC(GPP)5GAOGLRGGAGPOGPEGGKGAAGPOGPA(GPP)5GPC-NH2 | 45.3 |
| III30-A24-27 | GPC(GPP)5GAOGLRGGAGPOGPEGGKGAAGPAGPA(GPP)5GPC-NH2 | 40.8* |
| Peptide | Sequence | Tm (°C) |
|---|---|---|
| (GPO)5 | (GPO)5-NH2 | 28.2 |
| (GPO)3 | (GPP)2(GPO)3(GPP)2-NH2 | 41.1 |
| CRP | GCO(GPO)10GCOG-NH2 | 82.3 |
| III30 | GPC(GPP)5GAOGLRGGAGPOGPEGGKGAAGPOGPO(GPP)5GPC-NH2 | 47.4 |
| III30-A3 | GPC(GPP)5GAAGLRGGAGPOGPEGGKGAAGPOGPO(GPP)5GPC-NH2 | 39.0 |
| III30-A5 | GPC(GPP)5GAOGARGGAGPOGPEGGKGAAGPOGPO(GPP)5GPC-NH2 | 46.9 |
| III30-A6 | GPC(GPP)5GAOGLAGGAGPOGPEGGKGAAGPOGPO(GPP)5GPC-NH2 | 44.9* |
| III30-A8 | GPC(GPP)5GAOGLRGAAGPOGPEGGKGAAGPOGPO(GPP)5GPC-NH2 | 44.8* |
| III30-A11 | GPC(GPP)5GAOGLRGGAGAOGPEGGKGAAGPOGPO(GPP)5GPC-NH2 | 41.3 |
| III30-A12 | GPC(GPP)5GAOGLRGGAGPAGPEGGKGAAGPOGPO(GPP)5GPC-NH2 | 42.2 |
| III30-A14 | GPC(GPP)5GAOGLRGGAGPOGAEGGKGAAGPOGPO(GPP)5GPC-NH2 | 43.3 |
| III30-A15 | GPC(GPP)5GAOGLRGGAGPOGPAGGKGAAGPOGPO(GPP)5GPC-NH2 | 47.4 |
| III30-A17 | GPC(GPP)5GAOGLRGGAGPOGPEGAKGAAGPOGPO(GPP)5GPC-NH2 | 44.8* |
| III30-A18 | GPC(GPP)5GAOGLRGGAGPOGPEGGAGAAGPOGPO(GPP)5GPC-NH2 | 45.5 |
| III30-A23 | GPC(GPP)5GAOGLRGGAGPOGPEGGKGAAGAOGPO(GPP)5GPC-NH2 | 45.0 |
| III30-A24 | GPC(GPP)5GAOGLRGGAGPOGPEGGKGAAGPAGPO(GPP)5GPC-NH2 | 44.4 |
| III30-A26 | GPC(GPP)5GAOGLRGGAGPOGPEGGKGAAGPOGAO(GPP)5GPC-NH2 | 47.2 |
| III30-A27 | GPC(GPP)5GAOGLRGGAGPOGPEGGKGAAGPOGPA(GPP)5GPC-NH2 | 45.3 |
| III30-A24-27 | GPC(GPP)5GAOGLRGGAGPOGPEGGKGAAGPAGPA(GPP)5GPC-NH2 | 40.8* |
Tm was estimated by the method of Persikov et al.51
Collagen binding assay
Enzyme-linked immunosorbent assays (ELISAs) measuring the binding of dimeric GPVI-Fc mutants to human placental collagen type I (Sigma Aldrich) or collagen peptides were performed as described.22 Details of fibril preparation, binding measurements, and data processing are provided in the supplemental Methods.
Crystallization and data collection of GPVI alone and in complex with homotrimeric collagen peptides
Before crystallization, GPVI loop-truncation mutant ΔPAVS-ΔPAPYKN (10 mg/mL in gel-filtration buffer) was deglycosylated overnight at 292 K by addition of 1:100 (vol:vol) EndoHF (NEB; 1 × 106 unit/mL). GPVI crystals were obtained in approximately 1 month by sitting-drop vapor diffusion with well solution containing 0.1 M phosphate-citrate buffer; pH, 4.0; and 40% (vol:vol) PEG-300 at 292 K. Crystals were flash-frozen in liquid nitrogen without further cryoprotection. Diffraction data to 1.9 Å resolution were collected on the PX-beamline at the Swiss Light Source.
For cocrystallization of GPVI with collagen peptide (GPO)5 or (GPO)3 (Table 1), protein and peptide were mixed in a 1:1.2 molar ratio. Crystals of the GPVI-(GPO)5 complex were grown at 292 K with crystallization buffer containing 0.1 M MES; pH, 6.0; 0.2 M MgCl2; and 20% (wt:vol) PEG-6000. Crystals of the GPVI-(GPO)3 complex were grown with buffer containing 0.1 M MMT (DL-malic acid, MES monohydrate, Tris); pH, 9.0; and 25% (wt:vol) PEG-1500. Crystals were cryoprotected in crystallization buffer containing 20% (vol:vol) glycerol before flash-freezing in liquid nitrogen. Diffraction data with 2.5 Å resolution limits for both complexes were collected on the ID23-1 beamline at the European Synchrotron Radiation Facility. Details about data processing and structure solving are provided in the supplemental Methods.
Results
Crystal structure determination of GPVI collagen–peptide complexes
To obtain crystals of GPVI collagen–peptide complexes, it proved essential to truncate two loops in GPVI-D2 that are flexible, judged from the published crystal structure (PDB-ID: 2GI7) (supplemental Figure 1A).27 From eight mutants tested, we selected ΔPAVS-ΔPAPYKN that lacks residues 102-105 and 131-136 (supplemental Figure 1B-D), which showed the least decrease in expression yield (20%) and the greatest increase in thermal stability (2.5°C). Its CRP- and collagen-binding affinity are both reduced about five to sixfold (supplemental Figure 1D-E), measured using dimeric Fc fusions of the mutant and WT protein (supplemental Methods).
We obtained structures of the GPVI ΔPAVS-ΔPAPYKN mutant alone (1.9 Å resolution) (Figure 1A; supplemental Figure 2A-C) and in complex with collagen peptides (GPO)5 and (GPO)3 (both at 2.5 Å resolution) (Figure 1B-C; Table 1; supplemental Figure 3A-I; supplemental Table 2). The complexes with collagen peptides exhibit the same crystal form with two prominent structural features (supplemental Figure 4A-C): two GPVI molecules bind one triple-helical peptide with the same D1 domain region (Figure 1B-C; supplemental Figure 5A), and each of these molecules additionally forms back-to-back D2 interactions with a symmetry-related receptor–peptide complex. The truncated D2 loops have recently been implicated in a dimer-switch mechanism,33 but in both situations, they are located so that even the longer loops in the WT protein could not reach the collagen peptide. Therefore, the mildly reduced collagen binding of the mutant used for crystallization cannot directly be explained by the crystal structures.
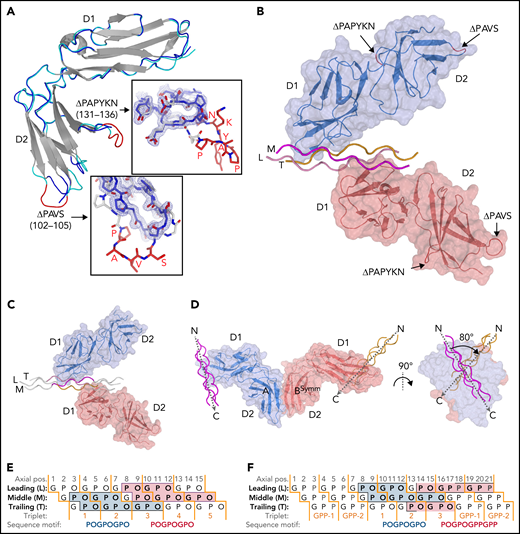
Crystal structures of GPVI mutant ΔPAVS-ΔPAPYKN and GPVI collagen peptide complexes. (A) Superimposition of the crystal structures of loop truncation mutant ΔPAVS-ΔPAPYKN (dark blue loops) and previously solved WT GPVI ectodomain (PDB-ID: 2GI7; light blue loops)27 showing significant conformational differences limited to the region around the truncated loops (red) in its D2 domain. The insets show an overlay of the WT loops (red) and the mutant loops (blue) in sticks representation, together with the 2Fo-Fc electron density map of the mutant (blue, 1.2σ contour level). See also supplemental Figures 1 and 2. (B) Crystal structure of the GPVI-(GPO)5 complex showing two GPVI molecules in cartoon and surface representations that bind the collagen peptide with their D1 domains. Chains of the collagen triple helix are shown in pink (leading; L), magenta (middle; M), and orange (trailing; T). Molecule A (blue) binds the M + T combination of chains; molecule B (salmon) binds the L + M chain combination. Also indicated (red) are the truncated D2 loops that are situated outside the region involved in collagen binding. See also supplemental Figure 3. (C) Crystal structure of the GPVI-(GPO)3 complex showing the same molecular arrangement as observed in crystals of the GPVI-(GPO)5 complex, but with shifted collagen chain stagger, such that the GPVI molecules bind the L + M and T + L combinations of chains, respectively. The two generic GPP repeats at either end of the (GPO)3 peptide are shown in white sticks. See also supplemental Figure 5. (D) Back-to-back D2–D2 interactions between two symmetry-related GPVI-(GPO)5 complexes. The interhelix angle of about 80° is incompatible with a parallel helix orientation as in collagen fibrils,34 and the back-to-back dimer is therefore not expected to be of functional relevance for in vivo binding to fibrillar collagen. See also supplemental Figure 4. (E-F) Schematic representation of the GPVI contact sites on the (GPO)5 (E) and (GPO)3 (F) peptides, also illustrating collagen chain stagger and GPO triplets. Collagen residues located within 4.5 Å of the first (blue) and second (salmon) GPVI molecule are highlighted, and the corresponding sequence motifs are depicted below the figure. Three out of 4 sites are formed by a POGPOGPO sequence; the fourth, the second site on (GPO)3, overlaps the generic C-terminal (GPP)2 extension of the peptide and is formed by the longer POGPOGPPGPP sequence due to the collagen chain stagger of the T + L chain combination.
Crystal structures of GPVI mutant ΔPAVS-ΔPAPYKN and GPVI collagen peptide complexes. (A) Superimposition of the crystal structures of loop truncation mutant ΔPAVS-ΔPAPYKN (dark blue loops) and previously solved WT GPVI ectodomain (PDB-ID: 2GI7; light blue loops)27 showing significant conformational differences limited to the region around the truncated loops (red) in its D2 domain. The insets show an overlay of the WT loops (red) and the mutant loops (blue) in sticks representation, together with the 2Fo-Fc electron density map of the mutant (blue, 1.2σ contour level). See also supplemental Figures 1 and 2. (B) Crystal structure of the GPVI-(GPO)5 complex showing two GPVI molecules in cartoon and surface representations that bind the collagen peptide with their D1 domains. Chains of the collagen triple helix are shown in pink (leading; L), magenta (middle; M), and orange (trailing; T). Molecule A (blue) binds the M + T combination of chains; molecule B (salmon) binds the L + M chain combination. Also indicated (red) are the truncated D2 loops that are situated outside the region involved in collagen binding. See also supplemental Figure 3. (C) Crystal structure of the GPVI-(GPO)3 complex showing the same molecular arrangement as observed in crystals of the GPVI-(GPO)5 complex, but with shifted collagen chain stagger, such that the GPVI molecules bind the L + M and T + L combinations of chains, respectively. The two generic GPP repeats at either end of the (GPO)3 peptide are shown in white sticks. See also supplemental Figure 5. (D) Back-to-back D2–D2 interactions between two symmetry-related GPVI-(GPO)5 complexes. The interhelix angle of about 80° is incompatible with a parallel helix orientation as in collagen fibrils,34 and the back-to-back dimer is therefore not expected to be of functional relevance for in vivo binding to fibrillar collagen. See also supplemental Figure 4. (E-F) Schematic representation of the GPVI contact sites on the (GPO)5 (E) and (GPO)3 (F) peptides, also illustrating collagen chain stagger and GPO triplets. Collagen residues located within 4.5 Å of the first (blue) and second (salmon) GPVI molecule are highlighted, and the corresponding sequence motifs are depicted below the figure. Three out of 4 sites are formed by a POGPOGPO sequence; the fourth, the second site on (GPO)3, overlaps the generic C-terminal (GPP)2 extension of the peptide and is formed by the longer POGPOGPPGPP sequence due to the collagen chain stagger of the T + L chain combination.
Similar interactions between back-to-back D2 domains have been observed in this and a previous report27 of collagen-free dimeric structures (supplemental Figure 4C); in the present collagen-bound form, the angle between collagen helices is large, about 80° (Figure 1D). The rigidity of this assembly impedes the reorientation of its D1 domains required to allow docking of the dimer onto the parallel helices that occur in fibrillar collagen.34,35 The two GPVI molecules that bind a single peptide make no contact with each other so that no cooperative interactions can favor the observed assembly. Instead, the absence of steric hindrance between GPVI monomers may permit this complex formation. Both structural features stabilize the observed crystal form, but neither would facilitate cooperative collagen binding in vivo.10,11
The GPVI binding site on collagen peptides
A GPVI binding site is comprised of two of the three collagen chains and has an area of about 470 Å2. In the GPVI-(GPO)5 complex, one GPVI molecule binds near the peptide N-terminus to a continuous surface comprising Pro2-Hyp6 and Pro2-Hyp9 residues of the middle (M) and trailing (T) chains, respectively; the other molecule binds to an equivalent site close to the C-terminus comprising residues Pro8-Hyp12 of the leading (L) chain and Pro8-Hyp15 of the M chain (Figure 1D). In the GPVI-(GPO)3 complex, one GPVI molecule binds to the L + M chains; a second to the T + L chains (Figure 1E). Interestingly, the latter site has a proline originating from the C-terminal (GPP)2 extension of the peptide (Table 1; Figure 1D) at a position that is occupied by hydroxyproline in the other (GPO)3 and both (GPO)5 binding sites. In these sites, the hydroxyl group does not contact GPVI, and therefore the presence of proline at this position likely does not influence binding.
The collagen-binding site of GPVI
The collagen-binding site of GPVI is located across the D1 β-sheet, comprising strands C, D, F, and F’ (Figure 2A). Its position differs from the collagen-binding site predicted from the crystal structure of GPVI alone,27 but is consistent with mapping of the collagen-binding site in LAIR-1.31 Eight central residues are either fully or highly conserved in GPVI orthologs (Figure 2B). Among them, Trp76 forms extensive hydrophobic interactions with T:Pro5 (one-letter prefixes L:, M:, and T: are used to refer to amino acids in the leading, middle, and trailing chain, respectively) and a hydrogen bond with the backbone carbonyl of T:Hyp3 (Figure 2C-E). Arg38 and Arg67 form hydrogen bonds with carbonyl oxygens of the (GPO)5 backbone. Gln71 and Glu40 are hydrogen bonded to the hydroxyl groups of M:Hyp3 and M:Hyp6, respectively (two occurrences of O in POGPOGPO) and appear essential for the hydroxyproline specificity of collagen substrates. The Tyr47 side chain is positioned close to M:Pro5 and M:Hyp6 and buries a substantial hydrophobic surface. Ser69 and Leu42 have hydrophobic interactions with T:Pro5 and T:Pro8, respectively. Four non-conserved residues, Leu36, Asp49, Ser74, and Gln82, provide minor interactions at the periphery of the binding interface. Finally, the Asn72 side chain and attached N-acetylglucosamine remaining after EndoH cleavage of its N-linked glycan are close to but oriented away from the collagen peptide (supplemental Figure 5B-C), and although its presence may contribute to the stability of the F-F’ loop, the N-glycan is unlikely to play a direct role in collagen binding.

The GPVI collagen-binding interface. (A) Surface representation of the GPVI collagen-binding site (magenta) with residues situated within 4.5 Å of collagen peptide (GPO)5 shown in sticks representation and β-strand labeled in green. (B) Representation of conservation within the GPVI family using ConSurf52 coloring illustrates strict conservation of six central and more variability in five surrounding residues in the collagen-binding site. (C) The GPVI-(GPO)5 interface showing hydrogen bonding interactions and salt bridges (dashed lines) and intermolecular carbon–carbon distances within 4.5 Å (thin solid lines). (GPO)5 residues in the leading (pink), middle (magenta), and trailing (orange) chain are shown in sticks representations and labeled by a sequence number (gray). Also displayed is the (GPO)5 surface, highlighting regions within 4.5 Å of GPVI by coloring according to the nearest chain. GPVI residues in the interface are shown as main chain ribbons and sidechain sticks (blue) and labeled by residue (black). (D-E) Detailed views of the hydrogen bonding interactions between (GPO)5 and GPVI residues Gln71 and Trp76 (D) and Arg38, Glu40, and Arg67 (E). Hydrogen bonds and salt bridges are indicated by dashed lines.
The GPVI collagen-binding interface. (A) Surface representation of the GPVI collagen-binding site (magenta) with residues situated within 4.5 Å of collagen peptide (GPO)5 shown in sticks representation and β-strand labeled in green. (B) Representation of conservation within the GPVI family using ConSurf52 coloring illustrates strict conservation of six central and more variability in five surrounding residues in the collagen-binding site. (C) The GPVI-(GPO)5 interface showing hydrogen bonding interactions and salt bridges (dashed lines) and intermolecular carbon–carbon distances within 4.5 Å (thin solid lines). (GPO)5 residues in the leading (pink), middle (magenta), and trailing (orange) chain are shown in sticks representations and labeled by a sequence number (gray). Also displayed is the (GPO)5 surface, highlighting regions within 4.5 Å of GPVI by coloring according to the nearest chain. GPVI residues in the interface are shown as main chain ribbons and sidechain sticks (blue) and labeled by residue (black). (D-E) Detailed views of the hydrogen bonding interactions between (GPO)5 and GPVI residues Gln71 and Trp76 (D) and Arg38, Glu40, and Arg67 (E). Hydrogen bonds and salt bridges are indicated by dashed lines.
Effect of GPVI mutations on collagen binding
The effect of mutations in the binding site was measured in solid-state assays using three collagen substrates that bind WT GPVI-Fc dimers with nanomolar affinity: fibrillar collagen I (KD = 30 nM), the synthetic agonist CRP (KD = 2.7 nM), and Toolkit peptide III-30 (KD = 11 nM). Binding to all three ligands is drastically reduced by nearly all alanine substitutions of conserved collagen-binding residues (Figure 3A-C; Table 2). Profound reductions were observed for mutants of Trp76, Arg38, and Glu40, showing >600-fold increase in KD relative to WT protein. Moderate reductions in affinity (17- to 360-fold) were observed for the Arg67, Tyr47, Ser69, and Asp49 mutants. Asp49 itself has little direct contact with collagen but, by forming a salt bridge, appears to fix Arg38 for optimal interaction, thus explaining the substantial effect of Asp49 mutation. In comparison with these substantial effects, alteration of residues inside the previously suggested C’-E groove27 affected collagen binding negligibly (supplemental Figure 6; supplemental Table 3), confirming that the crystal structures of the GPVI collagen complexes reveal the authentic collagen-binding site.

Mutational analysis of GPVI collagen binding. Binding of GPVI mutants (colored series) to (A) CRP, (B) fibrillar collagen I, and (C) Toolkit-peptide III-30 as measured in solid-state binding assays. Data measured as A450 is normalized to the binding of WT GPVI (black series). All data points represent the mean ± SD of at least 3 independent experiments. Binding curves are fitted to the equation Abs = (Bmax × c)/(KD,app + c) using nonlinear regression in SigmaPlot, where c is the GPVI concentration (μM). See also supplemental Figure 6.
Mutational analysis of GPVI collagen binding. Binding of GPVI mutants (colored series) to (A) CRP, (B) fibrillar collagen I, and (C) Toolkit-peptide III-30 as measured in solid-state binding assays. Data measured as A450 is normalized to the binding of WT GPVI (black series). All data points represent the mean ± SD of at least 3 independent experiments. Binding curves are fitted to the equation Abs = (Bmax × c)/(KD,app + c) using nonlinear regression in SigmaPlot, where c is the GPVI concentration (μM). See also supplemental Figure 6.
KD,app binding constants measured for WT and mutant GPVI-Fc binding to collagenous substrates in solid-phase plate-binding assays
| Mutant | CRP | Fibrillar collagen I | Toolkit peptide III-30 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| KD,app (nM) | 95% CI (nM) | Fold increase | KD,app (nM) | 95% CI (nM) | Fold increase | KD,app (nM) | 95% CI (nM) | Fold increase | |
| WT | 2.71 | 2.2-3.2 | 30.1 | 26-34 | 10.9 | 9.7-12.0 | |||
| E40A | 2.5 × 103 | 2.1-2.8 × 103 | 913 | 25 × 103 | 22.1-27.5 × 103 | 823 | 74 × 103 | 57-91 × 103 | 6848 |
| R38A | 2.0 × 103 | 1.7-2.3 × 103 | 748 | 18 × 103 | 16.0-20.3 × 103 | 603 | 42 × 103 | 38-45 × 103 | 3833 |
| W76A | 2.0 × 103 | 1.8-2.2 × 103 | 744 | 43 × 103 | 37.2-49.6 × 103 | 1440 | 21 × 103 | 14-27 × 103 | 1905 |
| D49A | 599 | 454-744 | 221 | 9.2 × 103 | 8.03-10.4 × 103 | 306 | 3.9 × 103 | 3.55-4.19 × 103 | 357 |
| R67A | 238 | 187-289 | 88 | 10 × 103 | 6.63-12.9 × 103 | 324 | 2.0 × 103 | 1.90-2.16 × 103 | 187 |
| Y47A | 160 | 130-189 | 59 | 6.0 × 103 | 4.43-7.56 × 103 | 199 | 10 × 103 | 8.5-12 × 103 | 925 |
| S69A | 45.7 | 24-67 | 17 | 1.2 × 103 | 0.93-1.49 × 103 | 40 | 6.5 × 103 | 5.66-7.40 × 103 | 602 |
| Q71A | 19.9 | 11-29 | 7 | 72.2 | 54.8-89.5 | 2 | 9.15* | 6.87-11.4 | 0.84 |
| L36A | 18.8 | 12-25 | 7 | 186 | 150-222 | 6 | 7.68* | 5.77-9.58 | 0.70 |
| Q82A | 3.89 | 2.5-5.3 | 1.4 | 433 | 329-538 | 14 | 289 | 268-310 | 27 |
| Mutant | CRP | Fibrillar collagen I | Toolkit peptide III-30 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| KD,app (nM) | 95% CI (nM) | Fold increase | KD,app (nM) | 95% CI (nM) | Fold increase | KD,app (nM) | 95% CI (nM) | Fold increase | |
| WT | 2.71 | 2.2-3.2 | 30.1 | 26-34 | 10.9 | 9.7-12.0 | |||
| E40A | 2.5 × 103 | 2.1-2.8 × 103 | 913 | 25 × 103 | 22.1-27.5 × 103 | 823 | 74 × 103 | 57-91 × 103 | 6848 |
| R38A | 2.0 × 103 | 1.7-2.3 × 103 | 748 | 18 × 103 | 16.0-20.3 × 103 | 603 | 42 × 103 | 38-45 × 103 | 3833 |
| W76A | 2.0 × 103 | 1.8-2.2 × 103 | 744 | 43 × 103 | 37.2-49.6 × 103 | 1440 | 21 × 103 | 14-27 × 103 | 1905 |
| D49A | 599 | 454-744 | 221 | 9.2 × 103 | 8.03-10.4 × 103 | 306 | 3.9 × 103 | 3.55-4.19 × 103 | 357 |
| R67A | 238 | 187-289 | 88 | 10 × 103 | 6.63-12.9 × 103 | 324 | 2.0 × 103 | 1.90-2.16 × 103 | 187 |
| Y47A | 160 | 130-189 | 59 | 6.0 × 103 | 4.43-7.56 × 103 | 199 | 10 × 103 | 8.5-12 × 103 | 925 |
| S69A | 45.7 | 24-67 | 17 | 1.2 × 103 | 0.93-1.49 × 103 | 40 | 6.5 × 103 | 5.66-7.40 × 103 | 602 |
| Q71A | 19.9 | 11-29 | 7 | 72.2 | 54.8-89.5 | 2 | 9.15* | 6.87-11.4 | 0.84 |
| L36A | 18.8 | 12-25 | 7 | 186 | 150-222 | 6 | 7.68* | 5.77-9.58 | 0.70 |
| Q82A | 3.89 | 2.5-5.3 | 1.4 | 433 | 329-538 | 14 | 289 | 268-310 | 27 |
Maximum III-30-binding of the Q71A and L36A mutants is 109% (104-114%) compared with WT protein.
Small but markedly substrate-dependent effects were observed upon mutation of Leu36, Gln71, and Gln82. Consistent with its peripheral location and lack of interaction in our crystal structure, mutation of Gln82 had no effect on binding to CRP. It did, however, significantly reduce binding to collagen I (14-fold) and III-30 (27-fold). Apparently, binding to more complex substrates exposing residues other than hydroxyproline at the C-terminal periphery of the binding site does involve Gln82 interactions, albeit not very strong. Mutation of Gln71 and Leu36 at the opposite end of the binding site had no effect or even slightly improved binding to III-30, whereas they reduced binding to both collagen I (two and sixfold, respectively) and CRP (both sevenfold). The modest effect of these mutations even on CRP binding indicates that the observed Gln71-M:Hyp3 hydrogen bond and Leu36-M:Hyp3 hydrophobic interactions contribute little to substrate binding. In combination, the substrate-dependent effects of Leu36, Gln71, and Gln82 mutants reveal a role for sequence variation at the amino- and carboxy-terminal periphery of the binding site in finetuning binding affinity.
An OGPOGP motif with minor peripheral sequence variation forms the core GPVI binding site
The 8-residue POGPOGPO sequence that forms the contact site in our (GPO)5 GPVI crystals (d < 4.5 Å) (Figure 1D) occurs once in the fibrillar region of collagen III15; nevertheless, ≥710,22 Toolkit-III peptides bind GPVI significantly. GPVI binding can apparently be mediated by a shorter sequence and/or sequences with one or more amino acid substitutions, as was also suggested by the differential effect of several GPVI mutants on binding to different collagen substrates. Calculations using the FoldX Suite36 indicate that the first proline and last hydroxyproline of POGPOGPO contribute only 6% of the binding energy (Figure 4A-B; supplemental Figure 7A-D) and can be replaced by any other amino acid without causing steric clashes with GPVI (see supplemental Methods for details). The shorter OGPOGP motif occurs 11 times in collagen III, and its presence in Toolkit-III peptides shows a better correlation with published binding data10 than any other shorter sequence does (supplemental Figure 8A; supplemental Methods).
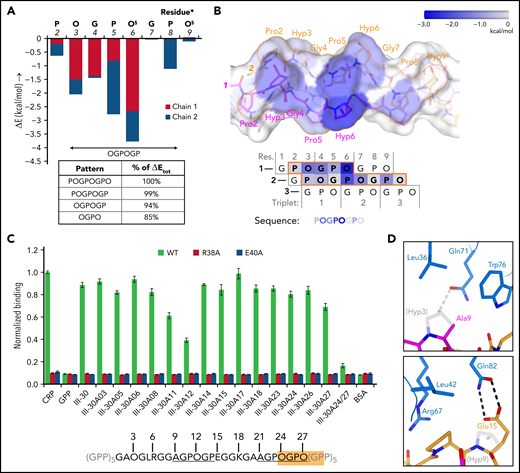
Binding pattern analysis of (GPO)5, (GPO)3, and Toolkit-peptide III-30. (A) Graph showing the relative contribution to GPVI binding energy by collagen–peptide residues calculated using the FoldX SequenceDetail function as in the supplemental Methods.36 Binding energy values were averaged for residues occupying equivalent positions in the 4 crystallographically resolved complexes; values for individual complexes are provided in supplemental Figure 7A-D; collagen residue numbering here conforms to Figure 2C. The inset table lists cumulative binding energies for different length sequence patterns and shown as percentage of the total energy. §Exists as Pro instead of Hyp in the second chain of the carboxy-terminal binding site of (GPO)3. (B) Surface rendering of (GPO)5 (top panel) and schematic representation of the chain stagger (bottom panel) displaying the calculated binding energy contributions per residue from panel A on a blue–white color scale. (C) Binding of WT GPVI-Fc (green) and control mutants R38A (red) and E40A (blue) to alanine-scanned variants of Toolkit peptide III-30 and control peptides III-30, CRP, (GPP)10, and bovine serum albumin (BSA). For easy reference, the amino acid sequence of III-30 is shown below, highlighting the presence of an OGPOGP motif (orange) and two AGPOGP motifs (underlined). Data measured as A450 is normalized to binding of WT GPVI to CRP. All data points represent the mean ± SD of at least three independent experiments. See also supplemental Figure 11. (D) Modeled binding of GPVI to the 9-AGPOGP-14 region of III-30. Relative to the (GPO)5 and (GPO)3 complexes, Ala instead of Hyp at position 9 disrupts interactions with GPVI Gln71 and Leu36 (left panel), whereas Glu instead of Hyp at position 15 just outside the motif enables the formation of an additional hydrogen bond to Gln82 (right panel). The predicted differences in interactions are in line with the divergent effect of GPVI mutations Gln71Ala, Leu36Ala, and Gln82Ala on binding to CRP and III-30 (see Figure 3A-C).
Binding pattern analysis of (GPO)5, (GPO)3, and Toolkit-peptide III-30. (A) Graph showing the relative contribution to GPVI binding energy by collagen–peptide residues calculated using the FoldX SequenceDetail function as in the supplemental Methods.36 Binding energy values were averaged for residues occupying equivalent positions in the 4 crystallographically resolved complexes; values for individual complexes are provided in supplemental Figure 7A-D; collagen residue numbering here conforms to Figure 2C. The inset table lists cumulative binding energies for different length sequence patterns and shown as percentage of the total energy. §Exists as Pro instead of Hyp in the second chain of the carboxy-terminal binding site of (GPO)3. (B) Surface rendering of (GPO)5 (top panel) and schematic representation of the chain stagger (bottom panel) displaying the calculated binding energy contributions per residue from panel A on a blue–white color scale. (C) Binding of WT GPVI-Fc (green) and control mutants R38A (red) and E40A (blue) to alanine-scanned variants of Toolkit peptide III-30 and control peptides III-30, CRP, (GPP)10, and bovine serum albumin (BSA). For easy reference, the amino acid sequence of III-30 is shown below, highlighting the presence of an OGPOGP motif (orange) and two AGPOGP motifs (underlined). Data measured as A450 is normalized to binding of WT GPVI to CRP. All data points represent the mean ± SD of at least three independent experiments. See also supplemental Figure 11. (D) Modeled binding of GPVI to the 9-AGPOGP-14 region of III-30. Relative to the (GPO)5 and (GPO)3 complexes, Ala instead of Hyp at position 9 disrupts interactions with GPVI Gln71 and Leu36 (left panel), whereas Glu instead of Hyp at position 15 just outside the motif enables the formation of an additional hydrogen bond to Gln82 (right panel). The predicted differences in interactions are in line with the divergent effect of GPVI mutations Gln71Ala, Leu36Ala, and Gln82Ala on binding to CRP and III-30 (see Figure 3A-C).
GPVI binding to sequence variants of OGPOGP with either of the terminal residues replaced by a residue equal to or smaller in size is evident (in hindsight) from previous studies10,22 (supplemental Figure 8B) and underpinned by our alanine-scanning study of the peptide of III-30 (Figure 4C; Table 1). We observed the strongest reduction of GPVI binding for single-residue mutants of Pro11 (-31%) and Hyp12 (-56%), proving the importance of the 9-AGPOGP-14 sequence for GPVI binding. Modeling of this site in the GPVI collagen complex reveals disruption of hydrogen bonding and hydrophobic interactions with GPVI residues Gln71 and Leu36 due to the Hyp-Ala replacement at the N-terminus of the motif (Figure 4D; supplemental Methods), in line with our previous observation that these GPVI residues do not contribute to III-30 binding (Figure 3C). Modeling also indicates that Glu15, located directly at the C-terminal of the AGPOGP-motif, can form an additional hydrogen bond to GPVI residue Gln82, a mutation of which markedly reduced binding to III-30 but did not affect CRP binding. These results demonstrate that the 6-residue OGPOGP motif implicated in high-affinity GPVI binding can tolerate conservative substitution of peripheral residues, whereas additional interactions with GPVI of residues just outside the core motif can further stabilize binding.
GPVI collagen binding is modulated by steric effects
The collagen fibril architecture34,35 and the D1–D2 interdomain angle of GPVI impose steric constraints that restrict the ability of GPVI to bind simultaneously to closely-spaced binding motifs in vivo (Figure 5A). At the collagen fibril surface, binding sites arising from identical motifs in parallel triple helices occur repetitively along the circumference of the fibril. Simultaneously binding to nearby sites on parallel helices <34 Å apart is prohibited by the bent shape and observed binding orientation of GPVI (Figure 5B). Binding to closely-spaced sites in the same triple helix as observed in our crystals is not feasible in vivo because the two GPVI molecules cover almost the entire helix circumference (Figure 5C; supplemental Figure 9A-B), whereas, in a collagen fibril, only a small part of the helix circumference is accessible to GPVI. Binding to the same side of the triple helix requires a much larger distance between GPVI molecules since its bent shape causes steric hindrance by D2. Depending on local collagen helicity, the minimum center-to-center distance between two OGPOGP binding sites should be 32 to 40 Å (ie, ≥2 triplets) (Figure 5D; supplemental Figure 10A-B; supplemental Methods). The shortest collagen fragment allowing association with two GPVI molecules simultaneously should therefore contain six triplets, longer than any naturally occurring GPO repeat, and thus must comprise discrete binding motifs.
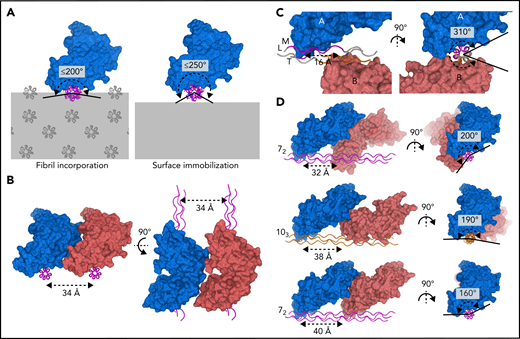
GPVI binding to fibrillar collagen requires a large distance between receptor molecules. (A) Schematic representations of GPVI binding (blue) to a triple helix (magenta) embedded in a collagen fibril (left panel) or immobilized onto a flat surface (right panel), illustrating that the circumferential angular range available for GPVI binding is about 200° and 250°, respectively. Additional GPVI molecules, covering about 160°, therefore need to bind at the same side of a helix or parallel helix. Triple-helical packing displayed in the left panel was taken from the in situ structure of collagen I, which exhibits a parallel helix packing leading to surface exposure of identical binding sites on parallel helices.34 (B) Perpendicular views of two GPVI molecules (blue and salmon) binding to identical sites on parallel helices (magenta) with minimum 34 Å spacing, showing its bent shape and oblique orientation with respect to the collagen helix preclude binding to closer-spaced helices. (C) Perpendicular views of the two GPVI molecules (blue and salmon) bound to (GPO)5 as observed in our crystal structure. A short center-to-center distance of only 16 Å (left panel) is accompanied by a large circumferential coverage of about 310° (right panel), which is not feasible if the helix is incorporated in the fibril or immobilized on a surface. See also supplemental Figure 9. (D) Perpendicular views of putative arrangements of two GPVI molecules (blue and salmon) bound to a triple helix in a 72 (top and bottom, magenta) or 103 (middle, orange) conformation. Their circumferential coverage angles are within the maximum value of 200° available for binding to fibrillar collagen; their center-to-center distance ranges from 32 to 40 Å, at least twofold larger than the distance observed in crystal structures. See also supplemental Figure 10 and supplemental Methods for details about modeling.
GPVI binding to fibrillar collagen requires a large distance between receptor molecules. (A) Schematic representations of GPVI binding (blue) to a triple helix (magenta) embedded in a collagen fibril (left panel) or immobilized onto a flat surface (right panel), illustrating that the circumferential angular range available for GPVI binding is about 200° and 250°, respectively. Additional GPVI molecules, covering about 160°, therefore need to bind at the same side of a helix or parallel helix. Triple-helical packing displayed in the left panel was taken from the in situ structure of collagen I, which exhibits a parallel helix packing leading to surface exposure of identical binding sites on parallel helices.34 (B) Perpendicular views of two GPVI molecules (blue and salmon) binding to identical sites on parallel helices (magenta) with minimum 34 Å spacing, showing its bent shape and oblique orientation with respect to the collagen helix preclude binding to closer-spaced helices. (C) Perpendicular views of the two GPVI molecules (blue and salmon) bound to (GPO)5 as observed in our crystal structure. A short center-to-center distance of only 16 Å (left panel) is accompanied by a large circumferential coverage of about 310° (right panel), which is not feasible if the helix is incorporated in the fibril or immobilized on a surface. See also supplemental Figure 9. (D) Perpendicular views of putative arrangements of two GPVI molecules (blue and salmon) bound to a triple helix in a 72 (top and bottom, magenta) or 103 (middle, orange) conformation. Their circumferential coverage angles are within the maximum value of 200° available for binding to fibrillar collagen; their center-to-center distance ranges from 32 to 40 Å, at least twofold larger than the distance observed in crystal structures. See also supplemental Figure 10 and supplemental Methods for details about modeling.
Our alanine-scanning study with Toolkit peptide III-30 reveals that solid-phase experiments employing randomly immobilized peptides closely mimic these inferred steric constraints (Figure 5A). Simultaneous knockout of the C-terminal 21-AGPOGP-26 and overlapping 24-OGPOGP-29 motif using a double Hyp24-Hyp27 substitution profoundly reduced GPVI binding (−81%) (Figure 3C). Single substitutions in either of these motifs, however, had only a minor effect (−4% to −22%). These results imply that exposed sites arising from both motifs can be occupied but not simultaneously (supplemental Figure 11A-C). Apparently, the overlapping motifs, despite being identical in length to the cocrystallized (GPO)3 motif, cannot associate with two GPVI molecules due to the steric constraints imposed by surface immobilization. The more N-terminal 9-AGPOGP-14 site binds GPVI independently, consistent with the conclusion above that binding sites should be ≥2 triplets apart (supplemental Figure 11D). Therefore, steric effects observed in GPVI-binding experiments to immobilized peptide III-30 are consistent with predictions for GPVI binding to fibrillar collagen.
Comparison of GPVI and LAIR collagen-binding sites
The previously mapped collagen-binding site of LAIR-1 is consistent with its position in GPVI, although its sequence is more variable within the LAIR family than within the GPVI family. The only residues strictly conserved within and between both groups are Trp76, Arg38, and Glu40 (GPVI numbering; supplemental Figure 12A-B). As for GPVI, mutation of these residues in LAIR-1 diminishes collagen binding drastically,31 and therefore, the Trp–Arg–Glu triad appears to be the core element in collagen binding by both receptor families. Additionally, GPVI residues Arg67, Tyr47, and Asp49 are conserved in human LAIR-1 and -2 but not in other species. In line with the extent of sequence divergence, the collagen-binding site of human GPVI more closely resembles crystal structures of human LAIR-1 (PDB-ID: 3KGR31) than mouse LAIR-1 (PDB-ID: 4ESK37) (supplemental Figure 12C-D). These structural differences and substantial divergences in Toolkit and CRP binding properties of both receptors32 underline the observation that peripheral sequence variation finetunes collagen-binding affinity and site-specificity.
Discussion
The platelet receptor GPVI binds to exposed collagen at sites of vascular injury, provoking downstream signaling and platelet aggregation,12 while inhibition or deficiency of GPVI protects from thrombus formation without severe bleeding disorders,38,39 indicating its potential value as a therapeutic drug target. Our crystal structures present the structural basis for GPVI binding to the canonical collagen sequence (GPO)n. In contrast to structures available to date of proteins in complex with collagen peptides, which all involve a collagen residue with a long side chain (ie, phenylalanine [VWF-A3,20 SPARC,17 and OSCAR21], glutamate [α2β140], or arginine [Hsp4741,42]), our structures reveal a novel collagen-binding mode employing the interaction of a rather flat surface across the D1 β-sheet of GPVI with several modestly-sized hydroxyprolines in collagen.
Combined with previous Toolkit-III binding studies,10,22 our data support OGPOGP as the template motif in collagen that permits strong in vitro binding by GPVI while allowing minor attenuating variations in its terminal positions. Such motifs, occurring in all collagens, are distributed throughout all five segments of the triple-helical domain of collagen III34 and appear to form mixed clusters of binding sites for GPVI and other receptors (supplemental Figure 13A-B). The III-22 OGPOGP and VWF-A3 motifs20 and the III-30 and integrin α2β1 motifs15 exemplify positioning of sites sequentially on the same helix. Fibrillar stacking34 also adds OGPOGP motifs of III-9, III-49, III-57, and the α2β1 GROGER motif to these clusters. Possibly, clustering of binding sites facilitates simultaneous receptor binding and interplay in vivo, as, for example, proposed for α2β1 and GPVI.43-45
The authenticity of in vivo GPVI binding sites in collagen III and heterotrimeric collagen I will be critically dependent on both the exposure of helix segments on the fiber surface at areas of the vascular lesion (reviewed in Herr and Farndale35) and the feasibility of concerted receptor binding to clustering sites. The overlapping III-30 sites (AGPOGP and OGPOGA in the native collagen III sequence) will allow binding of one GPVI molecule to the site supporting the strongest interactions and maximizing the total binding capacity of this region. Simultaneous binding of GPVI to the separate AGPOGP and of α2β1 to the nearby GMOGER15 appears structurally possible (supplemental Figure 13C). The frequent occurrence of GPVI binding sites complicates their authentication in vivo. On the other hand, the vital importance for collagen-induced signaling of the relatively sparse VWF- and α2β1-binding sites strongly supports the need for them to be accessible along with clustering GPVI sites, at least to the extent they are present on the same helix segment.
Collagen binding induces clustering11 or dimerization27 of platelet–surface GPVI, or, provided that GPVI exists partially as dimers before ligand binding,46,47 it provokes a shift in monomer–dimer equilibrium44 or perhaps a conformational switch to an activated dimer.33 Each additional platelet receptor molecule binding to collagen substantially strengthens GPVI-mediated adhesion through avidity effects: 100- to 900-fold stronger binding per molecule as estimated from monomeric GPVI and dimeric GPVI-Fc2 binding to collagen fibrils and peptides.10,48 In this context, fibril architecture34 with parallel receptor sites35 and parallel orientation of conjugated D1–D2 domains are both required; the D1–D2 angle is rigid in our and all other structural data.27,33 Dimers demonstrated to date crystallographically, including the recently reported nanobody-induced domain-swapped dimer (PDB-ID: 7NMU)33 (supplemental Figure 14), could not themselves bind parallel sites in collagen unless disrupted to allow the separate monomers the freedom to bind. The latter mechanism, however, is inconsistent with observations of increasing dimer levels upon collagen binding.10,11,44 Our study offers no indication of ectodomain dimerization suitable for cooperative collagen binding beyond avidity effects arising from clustering, but since steric hindrance dominates the regulation of GPVI collagen binding, GPVI clustering may require longer collagen helices than our peptides.49 It remains plausible that GPVI employs independent50 dimerization and collagen-induced activation mechanisms based on avidity alone.
Potential antiplatelet agents modulating GPVI-mediated platelet adhesion could inhibit association with collagen, downstream signaling via FcRγ, phosphorylation,4 or receptor dimerization50; the latter pending elucidation of its exact mechanism in platelet activation. Despite the substantial structural similarities with the LAIR-1 binding sequence, our structural identification of the authentic collagen-binding site and complementary collagen sequence may now support de novo development of specific agents targeting the collagen–GPVI interaction and minimizing drug crossreactivity between receptors (eg, GPVI and LAIR-1), which would be important for separating antithrombotic and immunomodulatory activities.
Acknowledgments
The authors thank the Swiss Light Source (Villigen, Switzerland) and the European Synchrotron Radiation Facility (Grenoble, France) for providing data collection facilities, and the beamline scientists for their help with data collection.
This work was supported in part by ECHO grant 700.58.006 from the Council of Chemical Sciences of the Netherlands Organization for Scientific Research to E.G.H. and Medical Research Council project grant G0400701 and Wellcome Trust grant 068724/Z/02/Z to R.W.F.
Authorship
Contribution: L.J.F., H.C.B., G.E.J., D.H., D.B., N.J., and M.V. performed research; L.J.F., H.C.B., G.E.J., R.W.F., and E.G.H. designed experiments; L.J.F., R.W.F., and E.G.H. wrote the manuscript; and R.W.F. and E.G.H. supervised the research.
Conflict-of-interest disclosure: R.W.F. is Chief Scientific Officer of CambCol Laboratories Ltd. The remaining authors declare no competing financial interests.
The current affiliation for R.W.F. is CambCol Laboratories Ltd, Littleport, Ely, Cambridgeshire, United Kingdom.
Correspondence: Richard W. Farndale, Department of Biochemistry, University of Cambridge, CB2 1QW, United Kingdom; e-mail: rwf10@cam.ac.uk.
Experimental X-ray diffraction data and PDB models for GPVI mutant ΔPAVS-PAPYKN and the GPVI-(GPO)5 and GPVI-(GPO)3 complexes have been deposited in the Protein Data Bank with PDB-IDs 5OU7, 5OU8, and 5OU9, respectively.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal