

Key Points
CTMCs in the blood of patients with SM are associated with more advanced disease, adverse prognostic features, and poorer outcome.

Abstract
Circulating tumor mast cells (CTMCs) have been identified in the blood of a small number of patients with advanced systemic mastocytosis (SM). However, data are limited about their frequency and prognostic impact in patients with MC activation syndrome (MCAS), cutaneous mastocytosis (CM) and nonadvanced SM. We investigated the presence of CTMCs and MC-committed CD34+ precursors in the blood of 214 patients with MCAS, CM, or SM using highly sensitive next-generation flow cytometry. CTMCs were detected at progressively lower counts in almost all patients with advanced SM (96%) and smoldering SM (SSM; 100%), nearly half of the patients (45%) with indolent SM (ISM), and a few patients (7%) with bone marrow (BM) mastocytosis but were systematically absent in patients with CM and MCAS (P < .0001). In contrast to CTMC counts, the number of MC-committed CD34+ precursors progressively decreased from MCAS, CM, and BM mastocytosis to ISM, SSM, and advanced SM (P < .0001). Clinically, the presence (and number) of CTMCs in blood of patients with SM in general and nonadvanced SM (ISM and BM mastocytosis) in particular was associated with more adverse features of the disease, poorer-risk prognostic subgroups as defined by the International Prognostic Scoring System for advanced SM (P < .0001) and the Global Prognostic Score for mastocytosis (P < .0001), and a significantly shortened progression-free survival (P < .0001) and overall survival (P = .01). On the basis of our results, CTMCs emerge as a novel candidate biomarker of disseminated disease in SM that is strongly associated with advanced SM and poorer prognosis in patients with ISM.
Introduction
Mast cell (MC) disorders encompass a heterogeneous group of diseases with highly variable clinical behavior and outcome, among which mastocytosis and (nonclonal) MC activation syndrome (MCAS) are the most prevalent entities.1,2 Mastocytosis on its own also consists of a heterogeneous disease that may or may not present with MCAS (eg, primary MCAS).3 Mastocytosis is typically characterized by the expansion and accumulation of clonal (eg, KIT-mutated) MCs in 1 or more organ systems, such as skin, bone marrow (BM), liver, spleen, and/or the gastrointestinal tract.1,4,5 The current World Health Organization (WHO) classification divides mastocytosis into variants restricted to a single tissue (ie, cutaneous mastocytosis [CM]) and systemic mastocytosis (SM) with simultaneous involvement of 2 or more organ systems (typically BM and skin) in addition to rare localized forms of mastocytosis (ie, MC sarcoma).6 SM is further subclassified by WHO into indolent SM (ISM, including the provisional variant of BM mastocytosis [BMM]), smoldering SM (SSM), and more advanced forms of SM consisting of aggressive SM, SM associated with another hematologic neoplasm (SM-AHN), and mast cell leukemia.7 In turn, MCAS includes a subset of mastocytosis and other clonal MCASs (ie, primary MCAS), together with a major group of secondary and idiopathic MCASs in which clonal MCs are not detected.8 Among patients with primary MCAS, those predominate who present with MCAS and who have the diagnostic criteria for ISM because of BM involvement by clonal mast cells (eg, CD25+ and KITD816V+) in the absence of skin lesions and involvement of other tissues together with other types of ISM and advanced SM. A small group of patients carry clonal MCs without fulfilling the diagnostic criteria for mastocytosis. These patients are grouped under the term monoclonal mast cell activation syndrome (MMAS). Of note, patients with either BMM or MMAS typically present with a very low clonal MC burden in BM.9
To reach different tissues and distant sites within a tissue, BM-derived MC precursors migrate via the blood to distant sites in BM and other tissues (eg, the skin and the gastrointestinal tract).10,11 Thus, circulating CD45intCD34+SSCloLin–CD117int/hiFcɛRI+ MC precursors, in the absence of more mature CD117hiCD45+CD34–FcɛRIhi MCs, are found at low counts in the blood of healthy adults.10,11 Likewise, low percentages of circulating MC precursors have also been reported in the blood of patients with SM at levels similar to those identified in healthy adults (median, ∼0.005% of all leukocytes).11 In SM, however, MC precursors might coexist in blood with KIT-mutated circulating tumor MCs (CTMCs) in at least a subset of patients.12,13
Thus, CTMCs have long been reported in patients with advanced SM on the basis of low-sensitive cytomorphologic and inmunophenotypic approaches, suggesting that the presence of CTMCs is a unique feature of more advanced and severe forms of the disease.14,15 More recently, Dahlin et al16 confirmed the presence of CD117hiCD34–FcɛRI+ CTMCs by flow cytometry in the blood of a limited number of patients with advanced SM (4 of 4; 2 with aggressive SM and 2 with SM-AHN); in contrast, they were unable to detect CTMCs in the blood of 6 patients with ISM. In parallel, Mayado et al12 have also detected CTMCs in the blood of ∼25% of patients with ISM who showed more adverse prognostic features, such as multilineage involvement of BM hematopoiesis by KITD816V, greater serum baseline typtase, and higher MC counts in BM.17-20 These findings suggest that CTMCs might also be present not only in patients with advanced SM but also in a fraction of patients with ISM in association with more adverse disease features. However, the actual frequency of patients with SM who have CTMCs and the potential impact of CTMCs on disease behavior and patient outcome still remain to be determined.
Here, we used a highly sensitive next-generation flow cytometry approach to investigate the frequency of CTMCs and CD34+ MC precursors in the blood of a large cohort of 214 patients with MCAS (n = 21), CM (n = 13), or SM (n = 180).21 Our ultimate goal was to determine the potential association between the presence of CTMCs in blood and the clinical behavior of these MC diseases and patient outcome.
Patients and methods
Patients and samples
Seven patients with MMAS and 14 patients with nonclonal MCAS (hereafter both are named MCAS) (57% males; median age, 53 years [range, 19-80 years]) and 193 patients with mastocytosis (51% males; median age, 57 years [range 10-85 years]) were studied in parallel with paired normal BM and peripheral blood samples from 14 healthy adults (64% men; median age, 60 years [range, 26-75 years]) who had no past or present history of allergy. In every patient, diagnosis and classification of MCAS were established according to Valent et al2 and diagnosis and classification of mastocytosis were established according to the WHO criteria.6 In SM-AHN, diagnosis of the AHN component was based on WHO criteria6,22 with the following distribution: myelodysplastic syndrome, 3 patients; myelodysplastic syndrome/myeloproliferative disorder, 4 patients; chronic eosinophilic leukemia/hypereosinophilic syndrome, 2 patients; plasma cell myeloma, 2 patients; and peripheral or mature T-cell leukemia, 1 patient. Patients were observed at the reference centers of the Spanish Network on Mastocytosis (REMA; Mast Cell Unit, Hospital Virgen del Valle, Toledo, Spain, and the Cancer Research Centre, Salamanca, Spain). Before entering the study, each patient gave their informed consent to participate according to the Declaration of Helsinki, and the study was approved by the local institutional ethics committees. Each patient with SM was further classified at diagnosis into the risk groups defined by the Global Prognostic Score for Mastocytosis (GPSM)23 for progression-free survival (PFS) and the International Prognostic Score for (Advanced) Mastocytosis (IPSM)24 for overall survival (OS). When this study closed, none of the patients with MCAS or CM had shown disease progression or death, whereas 19 patients with SM had progressed to more advanced forms of SM (supplemental Methods, available at the Blood Web site) after a median follow-up of 6 years (range, 1-31 years), and 5 patients with SM had died as a result of SM-related causes after a median follow-up of 7 years (range, 2-12 years). The main clinical, laboratory, and genetic features of patients with MCAS (n = 21), CM (n = 13), or SM (n = 180) are provided in supplemental Table 1.
Immunophenotypic studies
Blood and BM samples were separately collected in tubes containing (K3)EDTA and stained fresh (<24 hours after collection) with a combination of 8 different fluorochrome-conjugated markers (CD30, CD45, FcɛRI, CD25, CD33, CD117, CD203c, and CD34) (supplemental Table 2). Flow cytometry sample immunophenotyping, data acquisition, and analysis were performed for a total of 1 × 107 peripheral blood and >5 × 106 BM cells per tube following the recommendations of REMA, as described in detail in supplemental Methods. The presence of CTMCs was defined as a homogeneous population of ≥20 cells meeting previously established criteria25,26 for aberrant MCs (ie, SSCint/hiFSCint/hiCD25+CD30−/+CD45intCD117hiFcɛRIlow/+CD33+CD203c+) (Figure 1). In parallel, CD34+ hematopoietic precursor and stem cells (HPSCs) and their subset of MC-committed CD34+CD117++FcɛRI+CD203c+ precursors were also identified (Figure 1; supplemental Figure 1)11,12 and counted as described in supplemental Methods. Individual patients were subclassified on the basis of the overall phenotypic profile of BM MCs (BMMCs) into patients with a normal mature FcɛRI+CD117hiCD25– phenotype (n = 36), a mixed (coexisting) normal-mature FcɛRI+CD117hiCD25– and aberrant (mature and activated) CD25+ phenotype (n = 32), an aberrant mature FcɛRI+CD117hiCD25+ phenotype in the absence of normal BMMCs (n = 82), or an immature FcɛRI–/lowCD117+CD25+ BMMC phenotype (n = 55) (supplemental Methods).
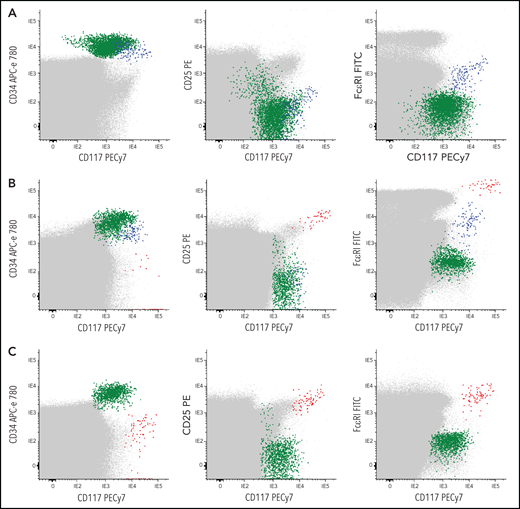
Representative bivariate dot plots illustrating the absence or presence of different levels of CTMCs and MC-committed CD34+ precursors in blood of representative CTMC–or CTMC+patients. (A-C) Data for whole blood cells are shown. Red dots represent CTMCs; blue dots, CD34+ MC precursors; green dots, other CD34+ cells; gray dots, other nucleated cells. (A) CTMC– and (B-C) CTMC+patients. APC, allophycocyanin; FITC; fluorescein isothiocyanate; PE, phycoerythrin.
Representative bivariate dot plots illustrating the absence or presence of different levels of CTMCs and MC-committed CD34+ precursors in blood of representative CTMC–or CTMC+patients. (A-C) Data for whole blood cells are shown. Red dots represent CTMCs; blue dots, CD34+ MC precursors; green dots, other CD34+ cells; gray dots, other nucleated cells. (A) CTMC– and (B-C) CTMC+patients. APC, allophycocyanin; FITC; fluorescein isothiocyanate; PE, phycoerythrin.
Gene mutation analyses
KITD816V was investigated in genomic DNA obtained from total blood and BM leukocytes, as well as fluorescence-activated cell sorted–purified (ie, >98% purity) populations of BM and blood MCs, neutrophils, monocytes, CD34+ HPSCs, and T cells using 2 previously described27,28 molecular methods (supplemental Methods) with a detection limit of 0.01%; the percentage of KITD816V-mutated cells was calculated using the delta-delta Ct (ΔΔCt) method, as previously described.28 On the basis of the results obtained with both methods, all patients were further subclassified according to the pattern of involvement of BM hematopoiesis by the KIT mutation as having either MC-restricted or multilineal involvement of BM hematopoiesis. In the former, the KIT mutation was restricted to BMMCs, and in the latter, it was also detected in other purified myeloid or myeloid plus lymphoid cell populations from BM.
Next-generation sequencing was performed in whole BM-derived genomic DNA from a subset of 65 patients with SM (6 with BMM, 38 with ISM, 5 with SSM, and 16 with advanced SM) for a total of 18 genes (ASXL1, CBL, CDH11, DNMT3A, EZH2, EPHA7, ICK, IKZF1, ITGA10, JAK2, KAT6B, KRAS, PIK3CD, ROS1, RUNX1, SF3B1, SRSF2, and TET2). In line with previous studies, SRSF2,ASXL1, and/or RUNX1 (S/A/R) gene panel, plus DNMT3A and EZH2 gene mutations, which have been associated with poorer patient outcome18,29,30 were used for further genetic subclassification of patients with SM. TET2, the most common gene mutated in SM was excluded from this subclassification because it showed no prognostic impact in SM.29,30
Statistical methods
Differences between groups were assessed by using the Kruskal-Wallis and the Mann-Whitney U tests (for continuous variables) or the χ2 test (for categorical variables), and correlation between variables was assessed by the two-sided Spearman’s rho (ρ) for nonparametric data. PFS and OS curves were plotted by the Kaplan-Meier method, and the statistical significance of differences between survival curves was estimated by the log-rank test. Statistical significance was set at P < .05 (supplemental Methods).
Results
CTMCs in the blood of patients with MCAS or mastocytosis
Overall, no CTMCs were detected in any of the healthy adults (0 [0%] of 14), patients with MCAS (0 [0%] of 21), or patients with CM (0 [0%] of 13) (Figure 2A). In contrast, CTMCs were found at relatively low numbers (median, 45 cells per mL) in almost half (83 [46%] of 180) of all patients with SM (Figure 2A; supplemental Table 3A). Among patients with SM, virtually all of those with advanced SM (24 [96%] of 25) or SSM (9 [100%] of 9) showed CTMCs in blood with median counts of 348 cells per mL and 24 cells per mL (P < .0001), respectively (Figure 2B; supplemental Table 3A). In turn, CTMCs were less frequently found in the blood of patients with ISM (47 [45%] of 105) and were rarely detected in patients with BMM (3 [7% of 41), where they were found at significantly (P < .0001) lower median CTMCs counts: 30 cells per mL in patients with BMM or ISM vs 348 cells per mL in patients with advanced SM (Figure 2C; supplemental Table 3A).

Distribution of CTMCs in blood of healthy donors and patients with MCAS, CM, or SM. Patients were classified according to (A-C) WHO diagnostic disease categories and (D) GPSM-PFS and (E) advanced SM (AdvSM)-IPSM risk stratification models. In each panel, boxes extend from the 25th to the 75th percentiles; the horizontal line in each box corresponds to the median value and vertical lines denote the 5th and 95th percentiles. In addition, the percentage of CTMC+ (ie, ≥10 CTMCs per mL) patients per diagnostic category is shown along the top of each panel. HD, healthy donor; Inter, intermediate; MCL, mast cell leukemia; PB, peripheral blood.
Distribution of CTMCs in blood of healthy donors and patients with MCAS, CM, or SM. Patients were classified according to (A-C) WHO diagnostic disease categories and (D) GPSM-PFS and (E) advanced SM (AdvSM)-IPSM risk stratification models. In each panel, boxes extend from the 25th to the 75th percentiles; the horizontal line in each box corresponds to the median value and vertical lines denote the 5th and 95th percentiles. In addition, the percentage of CTMC+ (ie, ≥10 CTMCs per mL) patients per diagnostic category is shown along the top of each panel. HD, healthy donor; Inter, intermediate; MCL, mast cell leukemia; PB, peripheral blood.
Of note, a significant association was observed between an immature BMMC phenotype and the presence of CTMCs in the blood (Figure 3A). Thus, almost every patient with advanced SM or SSM, as well as more than half of patients with BMM and patients with ISM who had CTMCs in the blood showed an immature (FcɛRI–/low) MC phenotype in BM (Figure 3A). In contrast, virtually all patients with BMM or ISM who had undetectable CTMCs in blood displayed a mature (FcɛRI+) BMMC phenotype (P < .0001), including a high frequency of patients with mixed phenotypically normal (CD25–) and aberrant (CD25+) mature MCs (46% and 26% of patients vs 0% in patients with SSM or advanced SM, respectively) (Figure 3A). In addition, BMMCs from patients with MCAS or CM systematically showed a normal (CD25–) mature (FcɛRI+) immunophenotype (Figure 3A). Conversely, CTMCs with a normal (CD25–) phenotype were not detected in any of the patients or healthy donors we investigated. In line with these findings, progressively lower expression levels of markers associated with mature MCs (ie, FcɛRI, CD203c, and CD33) were detected on the surface membrane of blood CTMCs from patients with BMM, ISM, or SSM (P = .015, P > .05, and P = .02, respectively) or advanced SM (P = .008, P < .0001, and P = .03, respectively) (Figure 3B-D). In contrast, similar levels of expression of CD117, CD25, and CD30 were observed on MCs from the distinct groups of SM in BM (data not shown) and in the blood (Figure 3E-G). Overall, CTMCs consisted of a population of CD34–CD117hiCD45+ MCs with either an (FcɛRI–/low) immature (CTMCimm) or (FcɛRI+/hi) mature (CTMCmat) but systematically aberrant (CD25+) phenotype, highly similar to that of BMMCs from the same patient (Figure 3A-G).
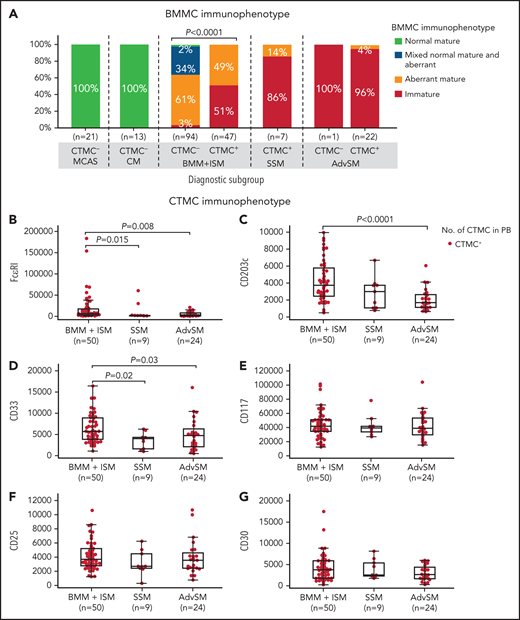
Most relevant phenotypic features of BMMCs and blood CTMCs in patients diagnosed with MCAS and distinct diagnostic categories of mastocytosis. On the basis of their BMMC phenotype, patients were classified as having a normal or reactive (CD25–CD2–CD30−/+CD33+FcɛRI+/hi) phenotype, a mixed normal (CD25–) and aberrant (CD25+) mature (FcɛRI+/hi) phenotype, an aberrant (CD25+) phenotype, or an immature (FcɛRI–/low) and aberrant (CD25+) phenotype. (A) Distribution of BMMC phenotypes according to diagnosis. (B-G) Levels of expression of distinct phenotypic markers on blood CTMCs expressed as mean fluorescence intensity (MFI) values (arbitrary units scaled from 0 to 262 144) per diagnostic category of SM are shown.
Most relevant phenotypic features of BMMCs and blood CTMCs in patients diagnosed with MCAS and distinct diagnostic categories of mastocytosis. On the basis of their BMMC phenotype, patients were classified as having a normal or reactive (CD25–CD2–CD30−/+CD33+FcɛRI+/hi) phenotype, a mixed normal (CD25–) and aberrant (CD25+) mature (FcɛRI+/hi) phenotype, an aberrant (CD25+) phenotype, or an immature (FcɛRI–/low) and aberrant (CD25+) phenotype. (A) Distribution of BMMC phenotypes according to diagnosis. (B-G) Levels of expression of distinct phenotypic markers on blood CTMCs expressed as mean fluorescence intensity (MFI) values (arbitrary units scaled from 0 to 262 144) per diagnostic category of SM are shown.
Circulating CD34+ MC precursors in the blood of patients with MCAS or mastocytosis
Similar median absolute counts of CD34+ precursors were found in the blood of healthy donors (1345 cells per mL), patients with MCAS (1155 cells per mL), and in patients with CM (1260 cells per mL) or SM (1435 cells per mL) (P > .05) (Figure 4A-C). In contrast, patients with MCAS or CM displayed higher absolute and relative numbers of MC-committed CD34+ precursors in blood compared with healthy donors (P ≤ .001 and P ≤ .006, respectively), whereas patients with SM had significantly lower median counts of MC-committed CD34+ precursors in blood (41 cells per mL) compared with patients with MCAS (113 cells per mL) or CM (98 cells per mL; P = .004 and P = .02, respectively) but not healthy controls (Figure 4D,G). Among patients with SM, MC-committed CD34+ precursors were particularly decreased toward undetectable levels (median, <0.0001 cells per mL) in SSM and advanced SM compared with BMM and ISM (60 cells per mL; P = .0003 and P < .0001, respectively) (Figure 4E,H). Likewise, lower absolute counts and percentages of MC-committed CD34+ precursors were also found among patients with BMM (P = .02) or ISM (P < .0001) with CTMCs in blood vs those who had undetectable CTMCs (Figure 4F,I). Altogether, these results translated into an inverse (nonlinear) correlation between the (absolute and relative) number of CTMCs in the blood of patients with MC-committed CD34+ precursors and CTMCs among patients with MCAS, CM, or SM (ρ = –0.57 and ρ = –0.58; P < .0001, respectively) (supplemental Figure 2A-B), when the analysis was also restricted to patients with mastocytosis who presented with multilineage involvement of BM hematopoiesis by KITD816V (P < .0001) (supplemental Figure 2C-D).

Distribution of total CD34+ HPC and MC-committed CD34+ precursors in the blood of patients with MCAS and different diagnostic categories of mastocytosis according to the absence or presence of CTMCs in blood. (A-C) HPSCs and (D-I) MC-committed CD34+ precursors in the blood of patients with MCAS or different diagnostic categories of mastocytosis, according to the absence or presence of CTMCs in blood. HPSCs, hematopoietic precursor and stem cells.
Distribution of total CD34+ HPC and MC-committed CD34+ precursors in the blood of patients with MCAS and different diagnostic categories of mastocytosis according to the absence or presence of CTMCs in blood. (A-C) HPSCs and (D-I) MC-committed CD34+ precursors in the blood of patients with MCAS or different diagnostic categories of mastocytosis, according to the absence or presence of CTMCs in blood. HPSCs, hematopoietic precursor and stem cells.
Clinical, laboratory, and genetic features of patients with SM who presented with CTMCs in their blood
The presence of CTMCs in the blood of patients with SM was associated with higher (median) age (62 vs 54 years; P = .0001) and female sex (58% vs 39%; P = .02), together with a greater frequency of skin lesions (77% vs 61%; P = .04) and organomegalies (46% vs 3%; P < .0001) and a lower rate of anaphylaxis (11% vs 41%; P < .0001; Table 1). In addition, patients with SM who had CTMCs in their blood also showed significantly lower (median) platelet counts (196 vs 223 × 109/L; P = .008) and hemoglobin levels (133 vs 144 g/L; P < .0001), lower total serum immunoglobulin E (5.4 vs 25.9 kU/L; P < .0001) and lactate dehydrogenase levels (152 vs 170 IU/L; P = .03), together with greater concentrations of serum baseline tryptase (108 vs 22.6 μg/L; P < .0001), serum alkaline phosphatase (99 vs 66 IU/L; P < .0001), β2-microglobulin (2.8 vs 2.0 μg/mL; P < .0001), and plasma histamine (0.13 vs 0.06 μg/dL; P < .0001) (Table 1). Of note, the presence of CTMCs in the blood of patients with SM was also associated with a higher frequency of diffuse bone sclerosis (15% vs 3%; P = .007) and greater percentages of BMMCs by flow cytometry (0.4% vs 0.09%; P < .0001) (Table 1). In line with this, significant (direct or inverse) correlations were found between CTMC counts in the blood and the above-mentioned blood and serum markers, even when the analysis was restricted to patients with SM who had CTMCs in their blood (supplemental Figure 3A-I).
Clinical, laboratory, and genetic features of patients with SM (n = 180) grouped according to the presence or absence of CTMCs in their blood
| Variable | CTMC–(<10 CTMCs per mL) (n = 97) | CTMC+(≥10 CTMCs per mL) (n = 83) | P | ||||||
|---|---|---|---|---|---|---|---|---|---|
| No./Total | % | Median | Range | No./Total | % | Median | Range | ||
| Clinical and laboratory features | |||||||||
| Age, y | 54 | 19-85 | 62 | 26-81 | .0001 | ||||
| Male sex | 59/97 | 61 | 35/83 | 42 | .02 | ||||
| Advanced SM * | 1/97 | 1 | 24/83 | 29 | <.0001 | ||||
| Anaphylaxis | 40/97 | 41 | 9/83 | 11 | <.0001 | ||||
| Skin lesions | 59/97 | 61 | 64/83 | 77 | .04 | ||||
| Organomegalies | 3/97 | 3 | 38/83 | 46 | <.0001 | ||||
| WBC × 109/L | 6.4 | 3.5-15.5 | 6.7 | 1.2-79 | .62 | ||||
| Hemoglobin, g/L | 144 | 106-176 | 133 | 86-168 | <.0001 | ||||
| Platelets × 109/L | 223 | 134-644 | 196 | 20-437 | .008 | ||||
| Serum baseline tryptase, μg/L | 22.6 | 2.2-485 | 108 | 17-1330 | <.0001 | ||||
| Serum lactate dehydrogenase, IU/L | 170 | 96-643 | 152 | 69-683 | .03 | ||||
| Serum alkaline phosphatase, IU/L | 66 | 38-146 | 99 | 35-990 | <.0001 | ||||
| Serum β2-microglobulin, μg/mL | 2.0 | 1.2-4.0 | 2.8 | 1.6-1.1 | <.0001 | ||||
| Plasma histamine, μg/dL | 0.06 | 0.01-2 | 0.13 | 0.04-1.1 | <.0001 | ||||
| Serum immunoglobulin E, kU/L | 25.9 | 1.9-1379 | 5.4 | 1.9-334 | <.0001 | ||||
| Loss of bone mass | 46/97 | 47 | 42/83 | 51 | .8 | ||||
| Bone sclerosis | 3/97 | 3 | 12/83 | 15 | .007 | ||||
| % BMMCs by flow cytometry | 0.09 | 0.002-0.9 | 0.4 | 0.02-37 | <.0001 | ||||
| Gene mutational profile | |||||||||
| % of patients with KITD816V mutation | 92/96 | 96 | 81/82 | 99 | .24 | ||||
| Multilineal KITD816V in BM† | 30/96 | 31 | 78/82 | 95 | <.0001 | ||||
| BM KIT VAF | <0.01 | <0.01-33.9 | 15 | <0.01-43.3 | <.0001 | ||||
| BM KITD816V+ cells ≥1% | 25/91 | 28 | 61/66 | 92 | <.0001 | ||||
| PB KIT VAF | <0.01 | <0.01-30.6 | 6.5 | <0.01-46 | <.0001 | ||||
| PB KITD816V+ cells ≥6% | 4/66 | 6 | 39/73 | 53 | <.0001 | ||||
| Pathogenic S/A/R/E/D mutations | 0/19 | 0 | 7/46 | 15 | .1 | ||||
| Disease progression | 0/97 | 0 | 19/83 | 23 | <.0001 | ||||
| Deaths | 0/97 | 0 | 5/83‡ | 6 | .02 | ||||
| Variable | CTMC–(<10 CTMCs per mL) (n = 97) | CTMC+(≥10 CTMCs per mL) (n = 83) | P | ||||||
|---|---|---|---|---|---|---|---|---|---|
| No./Total | % | Median | Range | No./Total | % | Median | Range | ||
| Clinical and laboratory features | |||||||||
| Age, y | 54 | 19-85 | 62 | 26-81 | .0001 | ||||
| Male sex | 59/97 | 61 | 35/83 | 42 | .02 | ||||
| Advanced SM * | 1/97 | 1 | 24/83 | 29 | <.0001 | ||||
| Anaphylaxis | 40/97 | 41 | 9/83 | 11 | <.0001 | ||||
| Skin lesions | 59/97 | 61 | 64/83 | 77 | .04 | ||||
| Organomegalies | 3/97 | 3 | 38/83 | 46 | <.0001 | ||||
| WBC × 109/L | 6.4 | 3.5-15.5 | 6.7 | 1.2-79 | .62 | ||||
| Hemoglobin, g/L | 144 | 106-176 | 133 | 86-168 | <.0001 | ||||
| Platelets × 109/L | 223 | 134-644 | 196 | 20-437 | .008 | ||||
| Serum baseline tryptase, μg/L | 22.6 | 2.2-485 | 108 | 17-1330 | <.0001 | ||||
| Serum lactate dehydrogenase, IU/L | 170 | 96-643 | 152 | 69-683 | .03 | ||||
| Serum alkaline phosphatase, IU/L | 66 | 38-146 | 99 | 35-990 | <.0001 | ||||
| Serum β2-microglobulin, μg/mL | 2.0 | 1.2-4.0 | 2.8 | 1.6-1.1 | <.0001 | ||||
| Plasma histamine, μg/dL | 0.06 | 0.01-2 | 0.13 | 0.04-1.1 | <.0001 | ||||
| Serum immunoglobulin E, kU/L | 25.9 | 1.9-1379 | 5.4 | 1.9-334 | <.0001 | ||||
| Loss of bone mass | 46/97 | 47 | 42/83 | 51 | .8 | ||||
| Bone sclerosis | 3/97 | 3 | 12/83 | 15 | .007 | ||||
| % BMMCs by flow cytometry | 0.09 | 0.002-0.9 | 0.4 | 0.02-37 | <.0001 | ||||
| Gene mutational profile | |||||||||
| % of patients with KITD816V mutation | 92/96 | 96 | 81/82 | 99 | .24 | ||||
| Multilineal KITD816V in BM† | 30/96 | 31 | 78/82 | 95 | <.0001 | ||||
| BM KIT VAF | <0.01 | <0.01-33.9 | 15 | <0.01-43.3 | <.0001 | ||||
| BM KITD816V+ cells ≥1% | 25/91 | 28 | 61/66 | 92 | <.0001 | ||||
| PB KIT VAF | <0.01 | <0.01-30.6 | 6.5 | <0.01-46 | <.0001 | ||||
| PB KITD816V+ cells ≥6% | 4/66 | 6 | 39/73 | 53 | <.0001 | ||||
| Pathogenic S/A/R/E/D mutations | 0/19 | 0 | 7/46 | 15 | .1 | ||||
| Disease progression | 0/97 | 0 | 19/83 | 23 | <.0001 | ||||
| Deaths | 0/97 | 0 | 5/83‡ | 6 | .02 | ||||
PB, peripheral blood; S/A/R/E/D, SRSF2, ASXL1, RUNX1, EZH2, and DNMT3A; VAF, variant allele frequency; WBC, white blood cell.
Eleven patients had ASM, 12 had SM-AHN, and 2 had mast cell leukemia.
One patient had the KITD816H mutation.
Three patients had ASM-AHN, 1 had ASM, and 1 had MCL.
From the genetic point of view, patients with SM who showed CTMCs in their blood more frequently displayed multilineage involvement of BM hematopoiesis by KITD816V (95% vs 31%; P < .0001), and they had higher KIT allele burden in blood (6.5 vs <0.01; P < .0001) and BM (15 vs <0.01; P < .0001) (Table 1). Interestingly, this translated into a significant (nonlinear) direct correlation between CTMC counts and the KITD816V allele burden in both blood (ρ = 0.70; P < .0001) and BM (ρ = 0.70; P < .0001) (supplemental Figure 4A-D), even when the analysis was restricted to patients with SM who had CTMCs in their blood (supplemental Figure 4A-B,D).
In addition to KITD816V, the presence of pathogenic mutations involving the SRSF2, ASXL1, RUNX1, EZH2, and DNMT3A (S/A/R/E/D) gene panel were also investigated in a subset of 65 patients with SM, 7 of whom (11%) showed ≥1 mutations (range, 1-3 mutations) in these latter genes. These included 5 (31%) of 16 patients with advanced SM (2 with SM-AHN, 2 with aggressive SM, and 1 with MC leukemia), 1 (20%) of 5 patients with SSM, and 1 (3%) of 38 patients with ISM. Noteworthy, all 7 (100%) of 7 patients with S/A/R/E/D mutations had CTMCs in the blood vs 20 (34%) of 59 patients with a wild-type phenotype (P = .1) (Table 1). Other mutations identified and their association with the presence of CTMCs in SM are listed in supplemental Table 4.
Association between the presence of CTMCs in the blood and outcome of SM
Although patients with and without CTMCs had been observed for similar periods of time since diagnosis, disease progression and deaths occurred only among patients with SM who were positive for CTMCs (23% and 6% vs 0% [P < .0001] and 0% [P = .02], respectively) (Table 1). To confirm the association between the presence of CTMCs in the blood and patient risk, we subsequently investigated the distribution of patients with SM who presented with vs without CTMCs in different risk groups defined by the GPSM-PFS and advanced SM-IPSM models (Figure 2D-E; supplemental Table 3B). Our results showed that only a small fraction of patients at low-risk of progression (22%) as defined by the GPSM-PFS score had CTMCs in the blood, with the percentage increasing to 55% among intermediate-risk patients and 95% among high-risk patients (P < .0001) (Figure 2D; supplemental Table 3B). Similarly, the frequency of patients with CTMCs in the blood progressively increased from low-risk (25%) and intermediate-1 risk patients (41%) to intermediate-2 (86%) and high-risk (100%) patients as defined by the advanced SM-IPSM model (P < .0001) (Figure 2E; supplemental Table 3B). In parallel, progressively higher absolute counts of CTMCs (P < .0001) were found in the blood of low- to intermediate-risk and high-risk patient groups as defined by the 2 score models (Figure 2D-E).
Survival analysis confirmed that the presence of CTMCs is an adverse prognostic factor associated with a significantly shortened PFS (median 10-year PFS, 80% [95% confidence interval (CI), 70%-90%] vs 100% [95% CI, 99%-100%]; P < .0001) and OS (median 10-year OS, 93% [95% CI, 87%-99%] vs 100% [95% CI, 99%-100%]; P = .01) (Figure 5A-B) in SM as a whole and when analysis was restricted to patients with nonadvanced SM (median 10-year PFS, 93% [95% CI, 85%-100%] vs 100% [95% CI, 99%-100%]; P = .008) (Figure 5C).

Impact of the presence of CTMCs in blood on the outcome of patients with mastocytosis. (A) PFS and (B) OS for the whole cohort of patients with mastocytosis (n = 193). (C) The impact of the presence of CTMCs on PFS of patients with nonadvanced SM (n = 155). NR, not reached.
Impact of the presence of CTMCs in blood on the outcome of patients with mastocytosis. (A) PFS and (B) OS for the whole cohort of patients with mastocytosis (n = 193). (C) The impact of the presence of CTMCs on PFS of patients with nonadvanced SM (n = 155). NR, not reached.
Discussion
CD34+ precursor cells in BM consist of a heterogeneous population of uncommitted and lineage-committed precursors, which includes a small fraction of early MC-committed CD34+ precursors.26,31,32 These CD34+ MC precursors migrate via the blood to different peripheral tissues where they complete maturation.10,33 Thus, circulating MC precursors with a CD34+CD117int/hiFcɛRI+ phenotype have been recurrently identified in the blood of virtually all healthy adults and patients with mastocytosis by sensitive flow cytometric approaches.10-13,26 In contrast to CD34+ MC precursors, early studies showed that the presence of CTMCs in blood was apparently restricted to patients with advanced SM, whereas they are absent in patients with ISM and in healthy participants.15,16,34 However, a recent (preliminary) study that used more sensitive flow cytometry techniques revealed CTMCs in the blood of 25% of patients with ISM who were studied at diagnosis.12 Such apparent discrepancy might be a result of the different phenotypic criteria used to define MCs and their degree of maturation, the variable sensitivities of the methods used to identify CTMCs in the blood of healthy donors and patients with SM, and/or the very limited number of patients investigated, among other reasons.12,15,16,35 Thus, the actual frequency and levels of both CTMCs and MC-committed CD34+ precursors present in the blood of patients with distinct diagnostic subtypes of mastocytosis and other MC disorders (eg, MCAS) remain largely unknown, as does their potential association with the clinical, biological, and prognostic features of the disease, particularly among patients with ISM, although they have not been previously investigated in MCAS and CM.
Here, we used a highly sensitive flow cytometry approach25 to determine the frequency and (absolute) counts of CTMCs and MC-committed CD34+ precursors in the blood of a large cohort of patients with MCAS, CM, and both nonadvanced SM (ie, BMM and ISM) and advanced SM. In line with previous observations by the REMA12 and other groups,15 our results confirmed that CD34+ precursors with an MC-committed (CD117hiFcɛRI+) phenotype are systematically present in the blood of patients with MCAS, CM, or SM and healthy donors. In contrast, phenotypically aberrant (CD34–CD117hiFcɛRI−/+CD25+) CTMCs were found in the blood of only a fraction of patients with SM, but they were absent in healthy donors as well as in all patients with MCAS or CM. Of note, circulating mature CD34–CD117hiFcεRI+CD25– MCs were undetectable in all patients with MCAS, CM, and SM and all healthy donors investigated here, including the few patients who had CD25– MCs in their BM. Among patients with SM, the frequency and absolute number of blood CTMCs progressively increased from <10% of patients with BMM to nearly half of patients with ISM and virtually all patients with SSM and advanced SM. Altogether, this accounts for the highest frequency of CTMCs noted so far in ISM, and to a lesser extent, also in BMM, and it almost doubles the percentage of patients with ISM who have CTMCs previously described in the literature.12 The high frequency of patients with CTMCs might be a result of the greater sensitivity of the method used here, which allows detection of down to 10 CTMCs per mL of blood compared with the level reached in previous studies (≤430 CTMCs per mL)12; however, the higher frequency of multilineage involvement of BM hematopoiesis by KITD816V found in our ISM series (>50%) vs previous ISM series (∼30%) might also contribute to our reported (increased) frequency of CTMCs in SM.19,20,36
In contrast to CTMCs, the number of MC-committed CD34+ precursors in the blood progressively decreased from MCAS, CM, and BMM to ISM, SSM, and advanced SM, leading to an inverse correlation with that of CTMCs. Of note, this association was independent from the total number of CD34+ precursors in the blood, because these latter cells did not show a significant association with the number of CTMCs and the diagnostic subtype of the disease. Altogether these results point out the coexistence of inverse migration kinetics in different diagnostic categories of MC diseases between BM-derived MC precursors with a normal phenotype and BM-derived phenotypically aberrant CD25+ CTMCs. Thus, in more advanced forms of mastocytosis, migration of BM-derived MC precursors to the blood seems to be progressively replaced by the release of more differentiated (tumor) MCs that tend to migrate to the circulation at disease stages characterized by higher BM infiltration levels, usually by phenotypically more immature (FcɛRI–/low) and aberrant (CD25+) MCs. These altered migration kinetics to the blood of MC precursors and CTMCs observed in SM might help explain the distinct patterns of tissue involvement that predominate in ISM (eg, BM and skin) vs advanced SM (eg, BM, blood, and lymphoid tissues, in the absence of skin lesions). In line with this hypothesis, patients with SM who had CTMCs in the blood more frequently showed organomegalies and cytopenias compared with patients with SM who had no CTMCs in the blood.
At present, the biological significance of the presence and levels of CTMCs and/or MC-committed CD34+ precursors in the blood of patients diagnosed with mastocytosis or MCAS still remains largely unknown. However, CTMCs identified in the blood of patients with SM systematically displayed aberrant CD25 expression in the context of a mature (FcɛRI+) or immature (FcɛRI–/low) MC phenotype, highly similar to that of paired BMMCs. Thus, immature CTMCs were typically found in patients with advanced SM and SSM who almost systematically presented with a phenotypically immature (FcɛRI–/low) BMMC phenotype, whereas mature CTMCs tended to also predominate in the blood of those patients with BMM or ISM who also displayed an immature MC phenotype in BM associated with multilineal involvement of BM hematopoiesis by the KITD816V mutation. In contrast, virtually all patients with BMM or ISM who had undetectable CTMCs in the blood showed a mature (FcɛRI+) MC phenotype in BM typically in association with an MC-restricted KIT mutation.
These observations point to a close relationship between CTMCs in the blood and tumoral BMMCs. In addition, a direct (nonlinear) correlation was also observed in our cohort between the number of CTMCs in the blood of patients with SM and the percentage of MCs in paired BM samples from the same individual. Altogether, these findings suggest a close association between a higher tumor burden, more immature BM and blood MC phenotypes, and higher CTMC counts in the blood. This would also translate to a greater potential for tumor dissemination, growth, and progression at multiple distant sites in BM and other (eg, lymphoid) tissues, which contrasts with the tissue migration profile of normal MC precursors.11,12 In line with this hypothesis, previous studies have demonstrated that the maturation profile of BMMCs in SM is closely associated with unique molecular and prognostic subtypes of the disease.26,37 In this regard, progressive accumulation of more immature tumor MCs in the BM of patients with mastocytosis would lead to a progressively higher release of aberrant (mostly immature) MCs and a parallel inhibition of the production and release of early MC-committed CD34+ precursors to the blood.12,25 Such altered MC kinetics and unique homing patterns of tumor vs normal MCs in BM and other tissues might contribute to a preferential tissue migration and infiltration by tumor MCs and thereby to a higher functional impairment of the tumor-infiltrated organs.12,15 In line with this hypothesis, our results confirm and extend previous observations showing that the presence of CTMCs in the blood of patients with mastocytosis is typically associated with more advanced forms of systemic disease.16,38 Thus, CTMCs were systematically absent in MCAS (ie, both MMAS and nonclonal MCAS), whereas progressively greater CTMC counts were found in the blood of an increasingly high proportion of patients with BMM or ISM, and virtually all patients with SSM or advanced SM. This also translated into more adverse disease features among patients with SM who presented with CTMCs in the blood, such as a greater frequency of organomegalies, increased percentages of BMMCs, lower hemoglobin levels, higher serum levels of β2-microglobulin, serum alkaline phosphatase, and serum baseline tryptase, a greater allele burden for the KIT mutation (in blood and BM), as well as a greater frequency of multilineage KITD816V mutation. This applied when the whole cohort of patients with SM was considered and also when analysis was restricted to patients with ISM. In addition, a close association was also observed between the presence of CTMCs in the blood and the prognostic risk groups of SM as defined by the GPSM-PFS and AdvSM-IPSM score models and patient outcome. Consequently, the presence of CTMCs in blood emerged as a powerful prognostic factor associated with a poorer PFS and OS in SM. Further studies in larger series of patients with SM with longer follow-up are required to confirm these findings, including the independent value of CTMCs to predict patient outcome.
Acknowledgments
We thank the Biobank at the Hospital Virgen de la Salud (BioB-HVS) No. B.0000520, Toledo, Spain, and Julie Chaccour for her assistance in editing the final English version of the manuscript.
This work was supported by grants from the Carlos III Health Institute co-financed by the European Regional Development Fund (PI19/01166), Asociación Española de Mastocitosis y Enfermedades Relacionadas (AEDM 2019), and Fondos de Investigación para Enfermedades Raras del Ministerio de Sanidad, Servicios Sociales e Igualdad. L.T.-R. was supported by a grant from Aid for the Promotion of Youth Employment and Implementation of the Youth Guarantee in R+D+i from the Ministerio de Ciencia, Innovación y Universidades (PEJ2018-003271-A).
Authorship
Contribution: A.H. performed experiments, analyzed the data, interpreted the results, created the figures, and helped write the article; J.I.M.-G. analyzed the data and critically reviewed the article; L.T.-R. performed the experiments; L.S.-M., A.P.-P., and A.C.G.-M. performed flow cytometry experiments and critically reviewed the article; A. Mayado recruited the controls, performed flow cytometry experiments, and critically reviewed the article; A. Matito recruited the patients and controls, performed clinical follow-up of the patients, and critically reviewed the article; M.J.-A. and C.C. performed KIT mutation experiments and analyzed and interpreted the results; I.A.-T. performed clinical follow-up of the patients, designed the research, supervised the study, and helped write the article; and A.O. designed the research, supervised the study, and helped write the article.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Alberto Orfao, Cancer Research Center, University of Salamanca, Paseo de la Universidad de Coimbra S/N, 37007 Salamanca, Spain; e-mail: orfao@usal.es.
Data sharing statement: Data for this study are available upon reasonable request by contacting Alberto Orfao via e-mail at orfao@usal.es.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal