


Abstract
Kaposi sarcoma (KS) herpesvirus (KSHV), also known as human herpesvirus 8, is the causal agent of KS but is also pathogenetically related to several lymphoproliferative disorders, including primary effusion lymphoma (PEL)/extracavitary (EC) PEL, KSHV-associated multicentric Castleman disease (MCD), KSHV+ diffuse large B-cell lymphoma, and germinotropic lymphoproliferative disorder. These different KSHV-associated diseases may co-occur and may have overlapping features. KSHV, similar to Epstein-Barr virus (EBV), is a lymphotropic gammaherpesvirus that is preferentially present in abnormal lymphoid proliferations occurring in immunecompromised individuals. Notably, both KSHV and EBV can infect and transform the same B cell, which is frequently seen in KSHV+ EBV+ PEL/EC-PEL. The mechanisms by which KSHV leads to lymphoproliferative disorders is thought to be related to the expression of a few transforming viral genes that can affect cellular proliferation and survival. There are critical differences between KSHV-MCD and PEL/EC-PEL, the 2 most common KSHV-associated lymphoid proliferations, including viral associations, patterns of viral gene expression, and cellular differentiation stage reflected by the phenotype and genotype of the infected abnormal B cells. Advances in treatment have improved outcomes, but mortality rates remain high. Our deepening understanding of KSHV biology, clinical features of KSHV-associated diseases, and newer clinical interventions should lead to improved and increasingly targeted therapeutic interventions.
Introduction
Only 3 viruses have been identified that cause hematologic malignancies in humans. The first was Epstein-Barr virus (EBV; also called human herpesvirus 4 [HHV4]), a gammaherpesvirus, identified in 1964 in endemic Burkitt lymphoma.1 This discovery was followed in 1980 with the identification of the causative agent of adult T-cell leukemia, the retrovirus human T-cell leukemia virus-1.2 In 1995, a third virus, Kaposi sarcoma (KS) herpesvirus (KSHV; also called HHV8), was found to be directly associated with primary effusion lymphoma (PEL),3 less than a year after its initial identification in KS.4 Soon, KSHV was also found in some cases of multicentric Castleman disease (MCD; KSHV-MCD), an atypical lymphoid proliferation.5 Since the discovery of KSHV, no additional viruses directly associated with human hematologic malignancies have been identified,6 although other viruses, including HIV and hepatitis C virus, likely contribute to hematologic disorders through indirect mechanisms.
Molecular pathogenesis
KSHV is transmitted largely through saliva and is acquired primarily in childhood in highly endemic areas or through sexual practices predominately between men.7 Consistent with its ability to induce B-cell lymphoproliferative disorders, it has been found in peripheral blood B cells from patients with KS.8 KSHV is also able to infect B cells in vitro, although with low efficiency. For poorly understood reasons, in contrast to EBV, this infection is not long lasting and does not lead to immortalization of the infected cells.9 KSHV has also been observed in B cells in lymph nodes from patients with HIV-reactive lymphadenopathies.10
Like other herpesviruses, KSHV is a double-stranded DNA virus that circularizes after cell entry and remains as an episome during latency, when only few KSHV-associated genes are expressed. During the lytic phase, nearly every viral gene (∼100 genes) is expressed, resulting in infectious virions and usually subsequent cell death. Evaluation of KSHV gene expression in cell lines and patient samples indicates that PEL tends to express latent genes, whereas at least a subset of infected cells in KSHV-MCD expresses both lytic and latent genes.11-13 Lytic gene expression results in higher KSHV viral loads in the blood, and because some of the lytic KSHV-associated genes encode for cytokines or induce cytokine production, this may account for many of the clinical manifestations seen in MCD and KSHV inflammatory cytokine syndrome (KICS).14
A number of viral proteins are expressed in PEL and KSHV-MCD, including the latency associated nulear antigen (LANA), viral interferon regulatory factor 3 (vIRF3; also called LANA2), and viral interleukin-6 (vIL-6), as demonstrated by immunohistochemistry.13,15 Other viral gene products, including viral cyclin (vCYC) and viral FLICE-inhibitory protein (vFLIP), are produced as well, because they use the same promoter and are present in the same transcripts as LANA, and have been confirmed to be expressed in PEL cell lines. This locus also encodes a number of microRNAs (miRs). A second set of latent transcripts in this region encodes for the kaposin proteins. These viral proteins play important roles in the pathogenesis of KSHV-associated B-cell lymphoid proliferations. Viral gene products with lymphomagenic properties expressed in PEL, as follows, are illustrated in Figure 1.
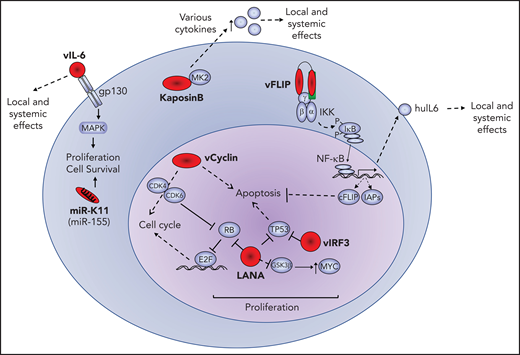
Oncogenic effects of viral oncoproteins. Latent viral proteins (LANA, vCYC, vFLIP, IRF3, and kaposin B) and messenger RNA (miR-K11) are expressed in PEL and depicted in red. Their effects or interactions with representative cellular proteins (blue) is shown. The pathogenic effects of these viral factors include increased cellular proliferation, inhibition of apoptosis and enhanced cellular survival, and production of human cytokines that may have local and systemic effects. cFLIP, cellular FLIP; huIL-6, human IL-6; IAP, inhibitor of apoptosis; MAPK, mitogen-activated protein kinase.
Oncogenic effects of viral oncoproteins. Latent viral proteins (LANA, vCYC, vFLIP, IRF3, and kaposin B) and messenger RNA (miR-K11) are expressed in PEL and depicted in red. Their effects or interactions with representative cellular proteins (blue) is shown. The pathogenic effects of these viral factors include increased cellular proliferation, inhibition of apoptosis and enhanced cellular survival, and production of human cytokines that may have local and systemic effects. cFLIP, cellular FLIP; huIL-6, human IL-6; IAP, inhibitor of apoptosis; MAPK, mitogen-activated protein kinase.
LANA
LANA (ORF73) is expressed in all KSHV-infected cells. LANA can be detected by commercial antibodies, which is helpful in the diagnosis of KSHV-associated diseases. LANA is highly immunogenic and therefore is useful in serologic assays detecting prior infection, which is helpful in understanding KSHV epidemiology. LANA plays an important role in viral replication, because it tethers the KSHV episomes to cellular chromosomes during cell division. It can also act as a transcriptional repressor, a function thought to be important for latency maintenance.16 LANA seems to contribute to lymphoid proliferation by dysregulating cell growth and survival, possibly by binding and inactivating the tumor suppressor proteins TP53 and retinoblastoma (RB1).17-19 LANA expression can also result in increased levels of MYC by binding to a negative regulator, GSK-3β.20,21
vCYC
vCYC (ORF72) is likely important in lymphomagenesis through its many functions. vCYC is a virally encoded cyclin D homolog that binds and activates cyclin-dependent kinase 6 (CDK6), inducing RB1 protein phosphorylation.22 This vCYC/CDK6 complex is less sensitive to inhibition by CDK inhibitory proteins than cellular cyclin D proteins and can induce S-phase entry and overcome RB1–mediated cell-cycle arrest triggered by CDK inhibitors. It also induces phosphorylation of p27 (CDKN1B), a key regulator of cell proliferation. vCYC overexpression induces apoptosis by inactivating BCL2,23 suggesting there are other mechanisms in KSHV-associated malignancies circumventing this effect. For example, it is possible that although TP53 mutations are rare in KSHV-associated malignancies, the functional inactivation of TP53 by LANA might allow vCYC to exert its effects. Other viral proteins, such as vFLIP and vIRF3, may also counteract the proapoptotic effects of vCYC. Furthermore, cellular homologs, such as cyclin D2 (CCND2), remain essential, perhaps by potentiating vCYC effects.24
vFLIP
vFLIP (ORFK13; ORF71) is the viral homolog of cellular FLIP (cFLIP), both of which contain 2 death effector domains. Whereas cFLIP can disrupt Fas-mediated death induction by competitive binding,25 vFLIP prevents apoptosis by upregulating NF-κB, which then induces expression of downstream antiapoptotic proteins and cytokines, including huIL-6.26,27 Inhibition of the NF-κB pathway and knockdown of vFLIP induces PEL cell death,28 indicating vFLIP plays a critical role in cell survival and may be a potential therapeutic target.29,30 vFLIP also is important in KSHV-MCD pathogenesis. vFLIP transgenic mouse models show an increased incidence of lymphadenopathy, with increased numbers of λ light chain–positive plasmablasts, similar to KSHV-MCD, as well as an increase in lymphoma, particularly in combination with MYC.31-33 Other effects of vFLIP include protection of cells from autophagy34 and impairment of lytic gene expression, thereby maintaining KSHV latency.
Kaposin B
Kaposin B is 1 of at least 3 proteins encoded in a genomic locus expressed in PEL cell lines during latency.35,36 It can bind and activate mitogen-activated protein kinase–associated protein kinase 2 (MK2, MAPKAPK2) as well as the upstream kinase, p38 mitogen-activated protein kinase (MAPK14).37 MK2 regulates the stability of cytoplasmic messenger RNAs that contain adenylate/uridylate-rich elements in their 3' untranslated regions. These adenylate/uridylate-rich elements are present in messenger RNA transcripts of cytokines (eg, IL-1, IL-3, IL-4, IL-6, tumor necrosis factor-α, and granulocyte-macrophage colony-stimulating factor), growth factors (eg, vascular endothelial growth factor), and oncogenes (eg, MYC). Thus, kaposin B, by upregulating MK2, prolongs the half-lives of these transcripts and increases cytokine production.37
vIL-6
vIL-6 is the viral homolog of huIL-6 and has been shown by immunohistochemistry to be expressed in variable proportions in KSHV+ lymphoproliferative lesions.38,39 Because it is a secreted cytokine, vIL-6 can have effects on neighboring cells, although intracellular vIL-6 seems to also promote proliferation and survival of PEL.40 vIL-6, like huIL-6, impairs B-cell apoptosis in response to apoptotic-inducing signals, thus likely playing a role in PEL and KSHV-MCD pathogenesis.41
vIRF3
vIRF3 (LANA2; ORFK10.5) is part of the IRF family. The vIFRs are homologs of cellular IRFs, which are transcription factors. One of these is cellular IRF4, which is critical for germinal center formation and important in B-cell differentiation, is highly expressed in cells with plasma cell differentiation, and, in PEL cells, is required for cell survival.24 It is possible that like IRF4, vIRF3 plays a role in B-cell differentiation and contributes to the plasma cell phenotype of PEL cells and KSHV-MCD plasmablasts.15,42,43 In addition, vIRF3 is a negative regulator of interferon responses.42,44 Furthermore, vIRF3 can prevent IRF5 from inducing TP53-independent apoptosis and p21-mediated cell-cycle arrest45 and independently binds and inactivates TP53,15 including by inhibiting sumoylation of TP53.46 These functions of vIRF3 could potentially compensate for the proapoptotic effects of vCYC, because studies of vIRF3 knockdown in PEL cells showed hampered cell proliferation and increased apoptosis.45
KSHV miRs
KSHV miRs arise from 12 pre-miR transcripts in the latency region, leading to the generation of at least 17 mature miRs.47-50 Publications on the effects of KSHV miRs are extensive and show that KSHV contributes to infected cell survival by inhibiting apoptosis and promoting viral latency.51 One of these in particular, KSHV-miR-K11, seems to have a potential lymphogenic effect. KSHV-miR-K11 is an ortholog of human miR-155,52 which has been implicated in B-cell lymphomagenesis53 and induces B-cell hyperproliferation in mouse models.54
Although KSHV encodes a remarkable number of viral genes with clear oncogenic functions, it does not transform B cells in culture. This makes KSHV different than EBV, which upon in vitro infection rapidly immortalizes B cells and allows the development of infected cell lines. EBV has a restricted latency pattern in doubly infected PEL, not expressing the major EBV oncoproteins,55 suggesting that this disease is largely driven by KSHV. However, EBV has also been found to be necessary for the proliferation of doubly infected PEL cells, consistent with the frequent coinfection by both viruses.56 So far, the best B-cell models for studying KSHV are PEL cell lines derived from patient specimens. The reason why KSHV is not transforming in current cell culture systems remains a mystery but may be due to its inability to establish stable latent infection in vitro.
KSHV-associated lymphoproliferations
Several KSHV-associated lymphoid entities in addition to PEL and KSHV-MCD have been described, including germinotropic lymphoproliferative disorder (GLPD), KSHV+ diffuse large B-cell lymphoma (DLBCL) not otherwise specified (KSHV-DLBCL), and extracavitary (EC) PEL (EC-PEL), but it seems these lesions are not as distinct as initially thought, with significant overlap now recognized. For example, it is sometimes difficult to separate EC-PEL from KSHV-DLBCL or GLPD from EC-PEL. Furthermore, KSHV+ cells can be seen in reactive lymph nodes without the classic morphologic features of MCD or in lymph nodes from asymptomatic HIV+ and HIV− individuals, a majority of whom were alive and well at follow-up (5-95 months).57-59 Thus, although in the following sections the current biologic, diagnostic, and therapeutic understanding of presently recognized KSHV-associated lymphoproliferative lesions will be discussed, future insights into KSHV biology and KSHV-associated proliferations may result in an evolution of their classification.
Because KSHV results in a systemic infection, it is important to evaluate any patient who is KSHV+ for KSHV-associated diseases, particularly if the patient is HIV+. KSHV-associated diseases frequently co-occur (Figure 2C-D). Therefore, it is critical for any KSHV+ patient with rapidly enlarging lymphadenopathy, an effusion, or clinical deterioration to have an extensive multidisciplinary clinical and laboratory evaluation, including positron emission tomography/computed tomography scans, tissue biopsies, effusion flow cytometry, and evaluation for opportunistic infections and other AIDS-related lymphomas.
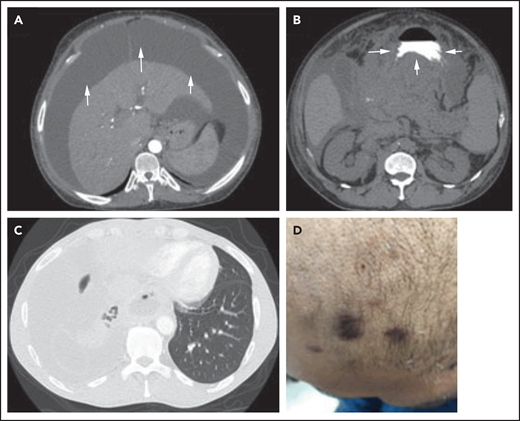
Three different presentations of PEL. (A) Isolated malignant ascites in a patient with classic PEL (arrows). (B) EC/solid PEL infiltrating the stomach (arrows). Notice the mass displacing the oral contrast. (C-D) Isolated malignant right-sided pleural effusion (C) with concurrent KS (D). Notice the collapse of the right lung compared with the left (C), with characteristic purple KS lesions located on the side of the face in the same patient (D).
Three different presentations of PEL. (A) Isolated malignant ascites in a patient with classic PEL (arrows). (B) EC/solid PEL infiltrating the stomach (arrows). Notice the mass displacing the oral contrast. (C-D) Isolated malignant right-sided pleural effusion (C) with concurrent KS (D). Notice the collapse of the right lung compared with the left (C), with characteristic purple KS lesions located on the side of the face in the same patient (D).
PEL
PEL is a rare, aggressive B-cell non-Hodgkin lymphoma defined by the presence of KSHV.3 PEL classically occurs in pleural, peritoneal, and pericardial spaces but can occur in other cavities, including blood vessels and the subarachnoid space.60-63 The solid EC variant of PEL usually arises in lymph nodes or in extranodal sites, particularly the gastrointestinal tract and skin (Figure 2B).
PEL/EC-PEL most frequently affects people living with HIV/AIDS (PLWHA), accounting for 2% to 4% of AIDS-related lymphomas, but also occurs in HIV− individuals, including elderly men from KSHV-endemic areas and solid organ transplant recipients.64-67 PEL/EC-PEL in PLWHA is usually both KSHV+ and EBV+ and occurs mostly in men with a median age of 42 years.62,65,66 HIV+ patients with PEL/EC-PEL usually present with CD4 counts <200 cells per μL, although the range is wide (Table 1).60,62,65,67-75 PEL/EC-PEL in HIV− individuals is infrequently EBV+. These patients are usually age 70 to 80 years at presentation and, in 1 case series, have had poorer outcomes.75
Patient characteristics and outcomes of clinical trials and case series of PEL
| Reference | ||||||||
|---|---|---|---|---|---|---|---|---|
| 93 | 71 | 68 | 65 | 62 | 72 | 74 | 73 | |
| No. of patients | 301 | 51 | 28 | 11 | 8 | 20 | 7 | 6 |
| Year of study | 2001-2012 | 1996-2013 | 1993-2003 | 1987-2002 | 1987-2001 | 2000-2013 | 2010-2017 | 2020 |
| PEL, % | 100 | 67 | 100 | 100 | 0 | 18 | 100 | 67 |
| Solid PEL, % | 24 | 33 | 0 | 0 | 100 | 1 | 0 | 33 |
| Median age, y | 55 | 45 | 44 | 41 | 40 | 44 | NA | 38 |
| HIV, % | 67 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| ART, % | 100 | 78 | NA | 20 | 100 | 100 | 100 | |
| Median (range) CD4+ T-cell count, cells per μL | NA | 204 (90-370) | 133 (5-756) | NA* | 125 (53-389) | NA | 231 (3-403) | |
| EBV, % | 50 | 66 | 71 | NA | NA | 73 | 60 | |
| KS, % | 28 | 49 | 67 | 27 | 25 | 75 | NA | 66 |
| MCD, % | 4 | 35 | 32 | NA | 30 | NA | 17 | |
| Receiving chemotherapy, % | 86 | 88† | 79 | 73 | 75 | 95‡ | 100 | 100 |
| Chemotherapy backbone | Various | CHOP-like | CHOP-like | CHOP | CHOP | DA-EPOCH | DA-EPOCH | DA-EPOCHR |
| CR, % | NA | 62 classic, 41 solid | 41 | 42 | NA | 53 | 71 | 50 |
| Median OS, mo | 6 | 10.2 | 6.2 | 6 | 11 | 22 | NA | Data not mature |
| OS rate, % | NA | 43 (classic) and 39 (EC) at 5y | 39 at 1y | NA | 40 at 5y | 47 cancer-specific survival at 3y | NA | 67 at 2y |
| EFS rate, % | NA | 71 (classic) and 100% (EC) DFS at 2y | NA | NA | NA | NA | 71 3y EFS | NA |
| Reference | ||||||||
|---|---|---|---|---|---|---|---|---|
| 93 | 71 | 68 | 65 | 62 | 72 | 74 | 73 | |
| No. of patients | 301 | 51 | 28 | 11 | 8 | 20 | 7 | 6 |
| Year of study | 2001-2012 | 1996-2013 | 1993-2003 | 1987-2002 | 1987-2001 | 2000-2013 | 2010-2017 | 2020 |
| PEL, % | 100 | 67 | 100 | 100 | 0 | 18 | 100 | 67 |
| Solid PEL, % | 24 | 33 | 0 | 0 | 100 | 1 | 0 | 33 |
| Median age, y | 55 | 45 | 44 | 41 | 40 | 44 | NA | 38 |
| HIV, % | 67 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| ART, % | 100 | 78 | NA | 20 | 100 | 100 | 100 | |
| Median (range) CD4+ T-cell count, cells per μL | NA | 204 (90-370) | 133 (5-756) | NA* | 125 (53-389) | NA | 231 (3-403) | |
| EBV, % | 50 | 66 | 71 | NA | NA | 73 | 60 | |
| KS, % | 28 | 49 | 67 | 27 | 25 | 75 | NA | 66 |
| MCD, % | 4 | 35 | 32 | NA | 30 | NA | 17 | |
| Receiving chemotherapy, % | 86 | 88† | 79 | 73 | 75 | 95‡ | 100 | 100 |
| Chemotherapy backbone | Various | CHOP-like | CHOP-like | CHOP | CHOP | DA-EPOCH | DA-EPOCH | DA-EPOCHR |
| CR, % | NA | 62 classic, 41 solid | 41 | 42 | NA | 53 | 71 | 50 |
| Median OS, mo | 6 | 10.2 | 6.2 | 6 | 11 | 22 | NA | Data not mature |
| OS rate, % | NA | 43 (classic) and 39 (EC) at 5y | 39 at 1y | NA | 40 at 5y | 47 cancer-specific survival at 3y | NA | 67 at 2y |
| EFS rate, % | NA | 71 (classic) and 100% (EC) DFS at 2y | NA | NA | NA | NA | 71 3y EFS | NA |
ART, antiretroviral therapy; CHOP, cyclophosphamide, doxorubicin, vincristine, and prednisone; CR, complete remission; da-EPOCH, dose-adjusted etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin; DA-EPOCHR, DA-EPOCH and rituximab; DFS, disease-free survival; EFS, event-free survival; NA, data not available; OS, overall survival; y, years.
Only 37% had data.
12 received low-dose/no treatment.
19 of 20 patients; 1 patient died before therapy.
Other lymphomas can present as lymphomatous effusions lacking KSHV, but these have different clinical and pathologic features than cases with KSHV, including older age at presentation, less frequent HIV positivity, common hepatitis C positivity, and frequent underlying medical conditions associated with fluid overload.76 Although the terms KSHV/HHV8− PEL and PEL-like lymphoma have been used in the literature, the consensus opinion of an international group of hematopathologists is that the designation of PEL should be restricted to cavity-based lymphomas with neoplastic KSHV-infected cells.77
The symptoms experienced by patients with PEL result from (1) the malignant effusion or EC mass, which, depending on its location, can lead to shortness of breath, abdominal distention, and chest pain (Figure 2A-C)62,65,67,68,71,75, and (2) KSHV and its associated conditions. Because KSHV is the tumor driver in PEL/EC-PEL, it is not uncommon for patients to present with other KSHV-associated malignancies/conditions (eg, KS or KSHV-MCD; Figure 1D; Table 1).62,65,67-69,71,75 One-third to 75% of patients with PEL present with concurrent or prior KS, and 33% have or have a history of KSHV-MCD (Figure 1D; Table 1). KSHV can also cause immune dysregulation, inducing elevations of huIL-10, huIL-6, and vIL-6, which can result in many of the constitutional and laboratory abnormalities, including fever, cachexia, edema, and anemia.72 Severe immune dysregulation can occur, leading to KICS, which in combination with other KSHV-associated conditions negatively affects prognosis.70,72
Pathologic diagnosis
PEL/EC-PEL cells are large and pleomorphic with immunoblastic/plasmablastic features. Cells that are Reed-Sternberg–like or anaplastic large-cell lymphoma-like are often seen (Figure 3A). By definition, PEL/EC-PEL is KSHV+ and uniformly expresses LANA (Figure 3A).3,60,62,71,75,77
![Histopathology of PEL and MCD. (A) PEL cells are large, with pleomorphic immunoblastic/plasmablastic features. A mitotic figure indicates proliferation of the tumor cells. Immunostaining of a cytospin preparation (insert) with an antibody to LANA shows the characteristic dot pattern in the nucleus (modified Giemsa stain, ×100 original magnification; insert: immunoperoxidase, ×100 original magnification). (B) Follicles in KSHV+ MCD are often involuted and hyalinized. There are many plasma cells in the interfollicular area and prominent vascular proliferation. In addition, a number of KSHV+ plasmablasts (arrows; insert) are present in mantle cell zones. These plasmablasts are LANA+ and vIL-6+ (hematoxylin and eosin [H&E] and immunoperoxidase staining, ×20 original magnification; inserts: ×60 original magnification).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/7/10.1182_blood.2020005470/5/m_bloodbld2020005470cf3.png?Expires=1769926182&Signature=IPj0Dppyz3dt386JpoZkElhXW-8LIAbfXdXXh6wwZ5sBWZRJV1sbEYoylT127DGHnF9xMvY0ALnXFxPYeEB4UkP3jv~BqFUWCjCPh1aDGamolT~9j2pbBSbpCDRsnzidJBkXABjKU5TIULd9rqXnY~aemChfVt0-IYVA9VRdQGjj40IY2g2C6qLlaCPRPKuid5fcCDRRPEuUkeqTgvW~8SHU7M5NQfArdu4q9KlvA5TShnYHm~RzGrFyn38JknERnMWm0bvArrDjnVXPMLCT18SsUO5cK8o7KCRyJBiE8EwKGKYe0niM40uJNcA3NjVbOxsocc0KbO6~9boP0EUdBw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Histopathology of PEL and MCD. (A) PEL cells are large, with pleomorphic immunoblastic/plasmablastic features. A mitotic figure indicates proliferation of the tumor cells. Immunostaining of a cytospin preparation (insert) with an antibody to LANA shows the characteristic dot pattern in the nucleus (modified Giemsa stain, ×100 original magnification; insert: immunoperoxidase, ×100 original magnification). (B) Follicles in KSHV+ MCD are often involuted and hyalinized. There are many plasma cells in the interfollicular area and prominent vascular proliferation. In addition, a number of KSHV+ plasmablasts (arrows; insert) are present in mantle cell zones. These plasmablasts are LANA+ and vIL-6+ (hematoxylin and eosin [H&E] and immunoperoxidase staining, ×20 original magnification; inserts: ×60 original magnification).
Histopathology of PEL and MCD. (A) PEL cells are large, with pleomorphic immunoblastic/plasmablastic features. A mitotic figure indicates proliferation of the tumor cells. Immunostaining of a cytospin preparation (insert) with an antibody to LANA shows the characteristic dot pattern in the nucleus (modified Giemsa stain, ×100 original magnification; insert: immunoperoxidase, ×100 original magnification). (B) Follicles in KSHV+ MCD are often involuted and hyalinized. There are many plasma cells in the interfollicular area and prominent vascular proliferation. In addition, a number of KSHV+ plasmablasts (arrows; insert) are present in mantle cell zones. These plasmablasts are LANA+ and vIL-6+ (hematoxylin and eosin [H&E] and immunoperoxidase staining, ×20 original magnification; inserts: ×60 original magnification).
A majority of PEL/EC-PEL cases lack markers of B-cell differentiation, such as CD19, CD20, CD22, CD79a, and PAX5, although a vast majority, by DNA analysis, are of B-cell origin. PEL/EC-PEL only occasionally expresses immunoglobulin with some λ and fewer κ light chain–positive cases described.3,60,62,71,75,78 The tumor cells also lack expression of germinal center markers (eg, CD10 and BCL6) but are positive for markers of plasma cell differentiation, including IRF4 (MUM1), BLIMP1 (PRDM1), CD38, and CD138, and activation-associated markers, such as CD30 (70% to 80% of cases) and epithelial membrane antigen. Sometimes PEL/EC-PEL exhibits aberrant T-cell antigen expression.71,77,78 However, T-cell antigen expression is not unique to KSHV+ EBV+ PEL/EC-PEL; it is also seen in KSHV− EBV+ lymphoma and KSHV+ EBV− PEL/EC-PEL.79-81 Rare cases are composed of monoclonal T cells, as evidenced by T-cell receptor rearrangements, and may be true cases of T-cell lymphoma.82 DNA analysis shows that monoclonal B cells in PEL/EC-PEL usually have somatic hypermutations in immunoglobin genes. Although mutations can be seen in protooncogenes, such as BCL6, MYC, PAX5, and RhoH/TTF, most PEL/EC-PEL cases do not contain mutations in the TP53 gene or rearrangements in common oncogenes or tumor suppressor genes such as MYC.3,60,62,78,83-86 An APOBEC mutational signature has been identified, consistent with viral infection.87 Gene expression studies show that PEL exhibits a distinct plasmablastic profile, intermediate between DLBCL and plasma cells, which is different from DLBCL in patients with and without HIV infection and more reminiscent of plasmablastic lymphoma.88,89 Thus, PEL/EC-PEL is likely derived from terminally differentiated B cells, which have likely gone through a germinal center reaction. EBV, if present, in PEL/EC-PEL usually shows a restricted gene expression pattern (latency I).55
Treatment and outcomes
In the largest cohort to date, comprising 51 patients (classic, n = 34; solid PEL, n = 17), 88% were treated with CHOP, with or without high-dose methotrexate, with concurrent ART. CR rate was 62%, and with a 10-year follow-up, median OS was 10 months (Table 1).71 In this study, there were no differences in survival based on the addition of methotrexate to CHOP, early ART vs current ART era, or EBV status. ART also seems to play an important role in the management of PEL. In this study, OS was almost doubled compared with CHOP-like treatment studies in the pre-ART/early ART era where ART was inconsistently or not administered either before or during treatment. In these earlier studies, CR rate was only 40% to 52%, with median OS of 3 to 6 months.60,65,68 In addition, PEL remission has been achieved using only ART. One retrospective study showed that no ART before PEL diagnosis was an independent poor prognostic factor.68 Thus, concurrent administration of ART with chemotherapy has become the current standard treatment of PEL/EC-PEL.90-92 Other poor prognostic factors identified in PEL/EC-PEL include Eastern Cooperative Oncology Group (ECOG) performance status >2, low CD4+ T-cell count, >1 body cavity involved by disease, and elevated lactate dehydrogenase.66,68,71,93
DA-EPOCH, an infusional chemotherapy, was investigated in 1 prospective study, with or without vorinostat, in an AIDS Malignancy Consortium trial (Table 1).74 This study evaluated treatment response in all AIDS-associated lymphomas, including 7 patients diagnosed with classic PEL. Vorinostat was specifically evaluated, because it is a potent histone deacetylase inhibitor that activates KSHV lytic gene expression and induces apoptosis in KSHV latently infected cells, such as PEL.69,74,94,95 Although the oncolytic ability of vorinostat to destroy KSHV/EBV-infected tumor cells was not established, the 7 patients treated with either DA-EPOCH (n = 3) or vorinostat + DA-EPOCH (n = 4) had a combined CR and 3-year event-free survival rate of 71%, constituting the most promising data to date (Table 1).74 In a retrospective study by Lurain et al,72 19 patients with HIV-associated classic PEL were studied, all of whom were treated with EPOCH backbone and concurrent ART. The patients had OS of 22 months and 3-year cancer-specific survival rate of 47%. Patients with EBV− PEL and/or elevated levels of IL-10 or IL-6 had poorer outcomes in this study.
KSHV-MCD
CD was originally described in 1956.96 In the 1980s, CD was divided into unicentric vs multicentric disease based on single vs multiple areas of lymphadenopathy.97,98 At the onset of the AIDS epidemic, an association between HIV and MCD was noted.99 Given the identification of KSHV in a large proportion of cases,5 MCD is now classified based on the presence or absence of this virus into (1) idiopathic (KSHV−) and (2) KSHV+ cases (ie, KSHV-MCD). KSHV-MCD cases are then subclassified based on HIV status, with a majority of cases being HIV+.100,101
Incidence of KSHV-MCD seems to have increased from the pre-ART to the current ART era.102 Median age at presentation is in the 40s in PLWHA and in the 60s in HIV− patients. Median CD4+ T-cell count at presentation in HIV+ patients ranges from 150 to 200 cells per μL. Multivariate analysis demonstrates that CD4+ T cells >200/μL, no ART exposure, and age >33 years are risk factors for developing KSHV-MCD.102,103
KSHV-MCD, like PEL, can present with other KSHV-associated malignancies. Fifty percent to 70% of patients with KSHV-MCD also have KS.102,103 Furthermore, HIV+ patients with KSHV-MCD have a 15-fold increase of developing non-Hodgkin lymphoma over the general HIV+ population.104 It seems that these patients are also more likely to develop KSHV+ lymphoma, as indicated in a study where 17 patients had non-Hodgkin lymphoma concurrently with KSHV-MCD and 16 had KSHV-associated lymphoma, including 9 patients with PEL and 7 with KSHV+ DLBCL.105
Patients with KSHV-MCD have an elevated KSHV viral load, evidence of viral replication, and symptoms of immune dysregulation accompanied by significant elevations in huIL-6, huIL-10, and vIL-6. These KSHV effects have been proposed to be important in the pathogenesis, the constitutional symptoms, and some of the laboratory abnormalities observed. Clinically, 80% to 100% of patients with KSHV-MCD present with fever, lymphadenopathy, and splenomegaly.106,107 Laboratory abnormalities include anemia, thrombocytopenia, and elevated erythrocyte sedimentation rate and C-reactive protein, in addition to the elevated cytokines.103,104,106,107 Therefore, during the workup before treatment, evaluation for other KSHV-associated conditions, HIV infection status, lymphoma, complete blood count, liver function, basic metabolic panel, and inflammatory markers (eg, IL-6, erythrocyte sedimentation rate, and C-reactive protein) is necessary to follow disease response during treatment.107 Articles by Abramson107 and Dispenzieri et al100 provide details on patient management beyond the scope of this review.
Pathologic diagnosis
KSHV-MCD lymph nodes show morphologic and immunophenotypic features similar to those of other types of CD. However, some KSHV-MCD lymph nodes may show features that overlap with those seen in HIV-related benign lymphadenopathy. Overall, the architecture of the lymph node is preserved with patent sinuses. There is usually prominent vascular proliferation in the interfollicular areas with significant numbers of plasma cells, which are polytypic by immunostaining. There may be a decreased number of interfollicular lymphocytes, particularly in HIV+ patients. The follicles are variably involuted and hyalinized. In some cases, there is a mixture of hyperplastic and involuted follicles. Prominent vessels penetrating follicles can be seen. The mantle cell zones may be pronounced, and the border between the mantle zone and the interfollicular area can be blurred.38,108-112
In contrast to other types of CD, medium to large relatively round mononuclear cells are seen (Figure 3B), which are the KSHV-infected cells first described as plasmablasts in 2000.109 Although this is likely a misnomer, KSHV+ cells in this disease have some features reminiscent of plasma cells, such as cytoplasmic immunoglobulin expression, MUM1 positivity, and relatively abundant cytoplasm. However, they differ from plasma cells in that they express B-cell antigens and are usually CD138−. These cells are preferentially seen in mantle cell zones and/or abutting germinal centers. Plasmablasts are positive for LANA; many also express cytoplasmic vIL-6 (Figure 3B). Expression of other KSHV lytic proteins by plasmablasts, including ORF59 and K8, has also been described. Plasmablasts are B cells that exhibit variable, often faint to negative, expression of CD20, CD79a, and PAX5 and lack expression of CD10 and BCL6, but they are uniformly positive for IRF4 and BLIMP1. Thus, these cells express markers of terminal B-cell differentiation. The cells are typically CD30− and CD138− but are usually CD38+.38,109,113-115 Plasmablasts also express the B-cell transcription factor OCT2, important for immunoglobulin expression. There is monotypic expression of immunoglobulin Mλ in LANA+ cells, with only a few random cytoplasmic κ+ cells seen in some cases. This latter finding may be due to the ability of KSHV to induce B-cell receptor revision via reinduction of Rag-mediated V(D)J recombination, with eventual switch from κ to λ light chain expression.116 KSHV+ plasmablasts are only rarely positive for EBV but show a high proliferation rate based on immunostaining for Ki67.38,77,109,110,112,113,115,117,118 The number of KSHV+ cells in a given case is variable, and in some cases, large aggregates of plasmablasts, previously known as microlymphomas, are seen. Molecular studies have found that most if not all cases of KSHV-MCD contain polyclonal or oligoclonal B cells, including cases with plasmablast aggregates. In addition, the cells comprising the plasmablast aggregates have unmutated immunoglobulin genes, indicating that they did not traverse the germinal center.115,119
Treatment and outcomes
Single- and multiagent cytotoxic chemotherapy regimens were studied initially for the treatment of KSHV-MCD, including CHOP, etoposide, and vinblastine. Although responses were rapid, their duration was not durable, and maintenance chemotherapy was sometimes used.103,106 In search for new treatment options, trials with rituximab, a monoclonal anti-CD20 antibody, were conducted with the idea that immunotherapy could destroy the KSHV-infected CD20+ plasmablasts, the potential disease driver.103,105,106,120-125 Two prospective trials assessed its efficacy as a single agent.120,121 In a study of 21 patients, single-agent rituximab was administered at 375 mg/m2per week for 4 weeks.120 Two-year OS and disease-free survival rates were 95% and 79%, respectively.120 Symptoms rapidly resolved, and 3 months posttherapy, only 10% had a positive KSHV viral load. However, 4 (36%) of 11 patients with concurrent KS demonstrated KS reactivation, despite concomitant ART.120,121 This KS flair was noted in other rituximab studies; therefore, single-agent rituximab should be avoided in KSHV-MCD with concurrent KS. Another benefit of single-agent rituximab therapy was a decreased incidence of lymphoma.105 A risk-adapted approach was also assessed. Patients with ECOG performance status <2 and no evidence of hemophagocytes, hemolytic anemia, or extranodal or splenic involvement were considered good risk and treated with weekly rituximab only for 4 weeks. High-risk patients were treated with concurrent etoposide.122,124 Eighty-four patients treated with this approach had 5-year OS and relapse-free survival rates of 92% and 82%, respectively,122 better than the 5-year OS rates ranging from 33% to 90% seen previously with standard chemotherapy without rituximab.124 Liposomal doxorubicin/rituximab was also studied prospectively in 17 patients with KSHV-MCD, resulting in 3-year OS rate of 81% and overcoming KS reactivation.125
Antiherpesviral drugs are known to inhibit lytic viral replication. However, single-agent antiviral therapy has not resulted in clinical benefit in KSHV-MCD. None of 7 patients responded to cidofovir, and only 2 of 4 patients with KSHV-MCD achieved remission with foscarnet.103 A study of high-dose valganciclovir and zidovudine was attempted, leveraging 2 KSHV lytic genes to phosphorylate zidovidine and valganciclovir into toxic agents. Although 12-month OS rate was 86%, CR rate was only 21%.126 Immunotherapy with anti–IL-6 antibodies has also been assessed; however, responses were not sustained, and CR rates were low.127,128
Currently, the standard treatment of KSHV-MCD remains rituximab in low-risk disease, with the addition of etoposide or pegylated liposomal doxorubicin (particularly in the presence of KS) for more active disease, all treated with concurrent ART in PLWHA.
As described, progression of KSHV-MCD to KSHV+ lymphoma has been documented. There is no current standard of care for the treatment of AIDS-related lymphoma in this setting. It should be noted, however, that improved rates of CR have been seen with infusional rituximab plus EPOCH, and in a retrospective study, superior OS was demonstrated with rituximab plus EPOCH over rituximab plus CHOP in KSHV− AIDS-related DLBCL.74,129
KICS
KICS is a recently described KSHV-associated hematologic condition, found initially in 6 HIV+ and KSHV+ patients who presented with an inflammatory syndrome similar to that seen in KSHV-MCD, but without biopsy-proven KSHV-MCD.130 Eighty percent of the patients had concurrent KS. Although the patients had no pathologic evidence of KSHV-MCD, all had huIL-6, huIL-10, and vIL-6 serum levels and KSHV viral loads similar to those of KSHV-MCD controls.130 In a subsequent series of 10 patients, median age was 36 years, all were HIV+, 80% were taking ART at diagnosis, and 50% had an undetectable HIV-1 viral load, with concurrent KS and/or PEL seen in 90% and 20% of patients, respectively.70 KICS has also been reported in KSHV+ HIV− patients after solid organ transplantation.131,132 The most common symptoms experienced by patients with KICS are fatigue, diarrhea, and edema.70 No patient had splenomegaly, and only 40% had lymphadenopathy, in contrast to patients with KSHV-MCD.70 Laboratory abnormalities were similar to those seen in patients with KSHV-MCD. Despite treatment of the concurrent KSHV-associated malignancy, median survival of patients with KICS was only 14 months, demonstrating the need for better therapy.70
Other hematologic diseases associated with KSHV
KSHV+ DLBCL
KSHV+ DLBCL not otherwise specified, previously known as large B-cell lymphoma arising in HHV8-associated MCD, is rare.133,134 These lesions often arise in the setting of KSHV-MCD, with cases that evolved from KSHV-MCD to KSHV-MCD with plasmablast aggregates to KSHV+ DLBCL.77,104,109,115,135,136 In contrast to aggregates of plasmablasts seen in KSHV-MCD, which disturb but do not obliterate the usual lymph node features, neoplastic plasmablasts in KSHV+ DLBCL form sheets and confluent clusters that destroy the underlying tissue architecture.77,104,109,115,135,136 The neoplastic cells are morphologically and immunophenotypically similar to KSHV+ plasmablasts in KSHV-MCD, exhibiting plasmablastic/immunoblastic features and expressing LANA, vIL-6, IRF4, and cytoplasmic immunoglobulin Mλ but lacking EBV. Some cases of KSHV-DLBCL show dim or focal expression of CD20 and/or CD79a, but many are negative for pan–B-cell antigens and are usually CD138−. Rare aberrant T-cell antigen expression has been described.77,104,109,115,135,136 DNA studies have shown that KSHV+ DLBCL is composed of monoclonal B cells that do not contain somatic hypermutations, consistent with a naïve B-cell derivation.104,109,115,136 KSHV+ DLBCL usually involves lymph nodes, spleen, and peripheral blood but can also be seen in extranodal sites.77,104,109,135,136
Outcomes in patients are very poor, ranging from weeks to months, especially in PLWHA, the most common presentation. This is markedly different from KSHV− AIDS-related DLBCL, where >75% of patients are alive at 3 years; therefore, there is a need for novel treatment approaches for these patients.74 However, the limited number of reported cases makes determination optimal therapy difficult.
GLPD
GLPD is a rare entity described in 2002.137 Only 19 cases have been described in the literature.138 It normally affects 50- to 60-year-old men who are mostly HIV− (78%), unlike other KSHV hematologic conditions, where immune suppression is an important hallmark.39,57,77,139 Clinically, all patients presented with nodal disease, and most are asymptomatic.138
These lesions are characterized by an infiltrate of pleomorphic KSHV+ EBV+ cells that partially or completely replace germinal centers with focal abnormal cells seen elsewhere in the lymph node. These cells do not disturb the overall lymph node architecture. Details of the morphologic, immunophenotypic, and genotypic findings were recently reviewed by Zanelli et al.138
Currently, there is no standard of care because of the paucity of cases.138 Chemotherapy alone was administered to 7 of 17 patients. Patients were treated with either CHOP or DA-EPOCH. Five of the 7 achieved remission (4-84 months), but 2 did not, even after undergoing autologous stem cell transplantation. Ten patients received no systemic therapy, with 5 receiving observation only, and the remaining patients undergoing either surgery or irradiation. In 2 patients who were observed, disease transformed to aggressive EBV+ DLBCL, 1 case of which was also KSHV+. Taken together, these data indicate this lymphoproliferative disorder seems to be indolent in nature and can be just observed, but it has the ability to transform to aggressive lymphoma and is quite responsive to chemotherapy.137-139
Future directions
Survival of patients with PEL has improved with DA-EPOCH, but outcomes remain poor, as a whole, in all KSHV-associated hematologic diseases. Targeted therapies based on disease biology and cellular vulnerabilities are likely to be identified in the coming years. Many preclinical studies have been performed in PEL cells, given the access to several cell lines. Some of these approaches have been explored clinically.
PEL has the disadvantage of having a null phenotype, and until recently, it was not amenable to targeted therapy. CD30, however, is present in 70% to 80% of PEL cases.75 The anti-CD30 antibody-drug conjugate brentuximab vedotin (BV) decreased cell proliferation, induced cell-cycle arrest, and triggered apoptosis of PEL cell lines.140 In vivo, BV promoted tumor regression and prolonged survival in xenograft PEL mice models. Single-agent BV has been used successfully in relapsed/refractory PEL to induce remission.141,142 CD38, commonly expressed in PEL, is another potential target. The anti-CD38 antibody daratumumab has also been used in relapsed/refractory PEL with success as monotherapy.143 KSHV-infected plasmablasts in KSHV-MCD are also strongly positive for CD38, much more so than CD20. Thus, daratumumab is an attractive potential treatment, either as a single agent or in combination with chemotherapy for KICS or KSHV-MCD. Because of their plasma cell differentiation, PEL cells are exquisitely sensitive to a nucleoside analog, 6-ehtylthioinosine, still in the preclinical stage. This is because plasma cells and other plasma cell malignancies express the enzyme adenosine kinase, which is required to activate this prodrug.144
Immunomodulatory drugs are also promising, because they decrease proliferation in most PEL cell lines.145 In vivo, lenalidomide downregulates IRF4 (MUM1), an essential protein overexpressed in PEL cell lines and tumor cells, and can reverse KSHV-induced downregulation of MCH-1 and ICAM-1.24,146 The efficacy of rituximab in the treatment of KSHV-MCD, along with the above data, provided the impetus for the design of an ongoing phase 1/2 trial of DA-EPOCH plus lenalinomide plus rituximab as upfront treatment of PEL (Table 1).73 The reported phase 1 portion of the phase 1/2 clinical trial demonstrated safety in the first 6 patients treated, with CR and 2-year OS rates of 50% and 67%, respectively (Table 1).73
Inhibiting viral gene products that affect KSHV-infected cell survival may add to our therapeutic arsenal in the future. One of these viral gene products is vFLIP, because knockdown of this protein in PEL cells results in a decrease in NF-κB and increased apoptosis. This is a difficult protein to target, being an adaptor protein, with its inhibition requiring disruption of a protein-protein interaction. As a proof of principle, this has been recently achieved through a peptide mimic of IKKγ (NEMO/IKBKG).30
Another potential viral target is miR-K11, which is an ortholog of miR-155, with an identical target sequence. An oligonucleotide inhibitor of miR-155 (cobomarsen) has been developed and tested in preclinical models and in a patient with DLBCL.147 Furthermore, a phase 2 clinical trial in cutaneous T-cell lymphoma using this drug was recently completed (registered at www.clinicaltrials.gov as #NCT03713320). The results of these studies may warrant evaluation of miR-155 inhibitors such as cobomarsen for potential targeting of the KSHV miR-K11 in PEL and KSHV-MCD. These targets and identification of future ones, with continued research on KSHV-associated conditions, are the keys to improving outcomes of patients with these aggressive KSHV-associated hematologic malignancies.
Acknowledgments
The authors apologize to all the authors they could not cite because of space constraints but whose contributions they greatly value.
E.C., A.C., and P.G.R. are partially supported by National Cancer Institute, National Institutes of Health grant 2UM1CA121947-14.
Authorship
Contribution: E.C., A.C., and P.G.R. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ethel Cesarman, Department of Pathology and Laboratory Medicine, Weill Cornell Medical College, New York, NY, 10065; e-mail: ecesarm@med.cornell.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal