

Key Points
Combining antimyeloma Id-KLH vaccine with vaccine-specific costimulated T cells led to significantly more robust IR.
Underlying immune suppression in patients with multiple myeloma could be overcome through combination with checkpoint inhibitor therapy.

Abstract
We hypothesized that combining adoptively transferred autologous T cells with a cancer vaccine strategy would enhance therapeutic efficacy by adding antimyeloma idiotype (Id)–keyhole limpet hemocyanin (KLH) vaccine to vaccine-specific costimulated T cells. In this randomized phase 2 trial, patients received either control (KLH only) or Id-KLH vaccine, autologous transplantation, vaccine-specific costimulated T cells expanded ex vivo, and 2 booster doses of assigned vaccine. In 36 patients (KLH, n = 20; Id-KLH, n = 16), no dose-limiting toxicity was seen. At last evaluation, 6 (30%) and 8 patients (50%) had achieved complete remission in KLH-only and Id-KLH arms, respectively (P = .22), and no difference in 3-year progression-free survival was observed (59% and 56%, respectively; P = .32). In a 594 Nanostring nCounter gene panel analyzed for immune reconstitution (IR), compared with patients receiving KLH only, there was a greater change in IR genes in T cells in those receiving Id-KLH relative to baseline. Specifically, upregulation of genes associated with activation, effector function induction, and memory CD8+ T-cell generation after Id-KLH but not after KLH control vaccination was observed. Similarly, in responding patients across both arms, upregulation of genes associated with T-cell activation was seen. At baseline, all patients had greater expression of CD8+ T-cell exhaustion markers. These changes were associated with functional Id-specific immune responses in a subset of patients receiving Id-KLH. In conclusion, in this combination immunotherapy approach, we observed significantly more robust IR in CD4+ and CD8+ T cells in the Id-KLH arm, supporting further investigation of vaccine and adoptive immunotherapy strategies. This trial was registered at www.clinicaltrials.gov as #NCT01426828.
Introduction
Over the last 2 decades, novel antimyeloma drugs such as proteasome inhibitors, immunomodulatory drugs, and monoclonal antibodies, in combination with autologous hematopoietic stem cell transplantation (auto-HCT), have improved outcomes in patients with multiple myeloma (MM). Despite these advances, MM remains an incurable cancer, with only a minority of patients achieving long-term disease control. Therefore, novel strategies are needed to prevent relapses and achieve durable responses.
Tumor immunotherapy holds great promise in MM, supported by the observation that allogeneic HCT can be potentially curative for a subset of patients because of the graft-versus-tumor effect. An active area of investigation in tumor immunotherapy has been the development of tumor-specific vaccines that can selectively target neoplastic cells while minimizing toxicities to the normal tissue. The variable region of the immunoglobulin molecules expressed on the surface of B cells and plasma cells contains unique determinants termed idiotypes (Ids) that can be recognized as tumor-specific antigens. B-cell lymphoma and MM involve clonal proliferation of neoplastic cells with a single unique Id. One randomized controlled clinical trial using a vaccine strategy targeting Id in follicular lymphoma positively demonstrated improved disease-free survival,1 although other trials have been conducted with mixed results.2-4 The setting in which a tumor vaccine is administered plays a crucial role in determining whether an effective immunological response can be induced, as well as in its clinical efficacy. Animal models have shown that, during the critical period of immune reconstitution after high-dose chemotherapy, there is a significant degree of tumor-specific T-cell activation, which can be further enhanced with tumor vaccines.5
Another recently successful approach in tumor immunotherapy has been the adoptive transfer of autologous immunocompetent cells for the treatment of hematological malignancies.6,7 The tumor cytoreduction and regulatory T-cell depletion achieved with high-dose chemotherapy and radiotherapy substantially increase the efficacy of adoptive immunotherapy.8,9 This makes auto-HCT an ideal platform for the combination of tumor vaccine and adoptive T-cell transfer to amplify the immune response and thereby increase the depth and durability of response. Several trials using this strategy have been conducted in MM with encouraging results.10-14
Our group previously conducted a randomized trial in follicular lymphoma using Id conjugated with a carrier protein, keyhole limpet hemocyanin (KLH), which showed superior disease-free survival with Id-KLH vaccination.1 We therefore designed this trial in patients with MM with the hypothesis that the Id-KLH vaccine plus vaccine-primed costimulated T cells would result in more robust Id-specific humoral and cellular responses compared with the control vaccine (KLH only).
Methods
Patient eligibility
This open-label randomized phase 2 trial was conducted at The University of Texas MD Anderson Cancer Center (Houston, TX) and at the University of Pennsylvania (Philadelphia, PA). Patients with newly diagnosed symptomatic MM with immunoglobulin G monoclonal protein, age ≤70 years, with at least stable disease within 10 months of induction therapy were included. Other inclusion criteria were Karnofsky performance score ≥80% and adequate cardiac, pulmonary, hepatic, and renal function. Patients were excluded if they had relapsed or progressive disease, had an uncontrolled infection, were HIV+, had an autoimmune disease other than Hashimoto’s thyroiditis, or had undergone a previous allogeneic or auto-HCT. All participants provided written informed consent in accordance with the Declaration of Helsinki; study approval was obtained from the institutional review boards of The University of Texas MD Anderson Cancer Center and the Abramson Cancer Center at the University of Pennsylvania and through an Investigational New Drug application accepted by the US Food and Drug Administration.
Trial design
The design of the trial is depicted in Figure 1. Eligible patients were randomly assigned 1:1 to either KLH or Id-KLH. Patients randomly assigned to the Id-KLH arm had an initial blood draw (100 cc) performed to obtain sufficient Id protein to prepare the Id-KLH vaccine, whereas patients in the KLH arm did not. Twenty-eight days before auto-HCT, those randomly assigned to KLH and Id-KLH arms received KLH and Id-KLH peptide vaccines, respectively. Granulocyte-macrophage colony-stimulating factor (GM-CSF; sargramostim) was injected subcutaneously as close as possible to the KLH-only or Id-KLH vaccine injection site. Two weeks before auto-HCT, all patients underwent steady-state apheresis to collect mononuclear cells for adoptive cellular therapy preparation. All patients received G-CSF mobilization and peripheral blood progenitor cell collection for auto-HCT. All patients were treated with high-dose melphalan (day −2) and underwent stem cell transplantation on day 0. Autologous lymphocytes were administered on day +2 to +5. Supportive care measures included antibiotic prophylaxis and use of G-CSF on day 5 until neutrophil count recovery to 1000/μL. Two additional sets of arm-specific immunizations were administered at days 30 and 90 after auto-HCT. There was no stratification of patients by prognostic variables. Immune response was evaluated by gene expression profiling using NanoString nCounter.
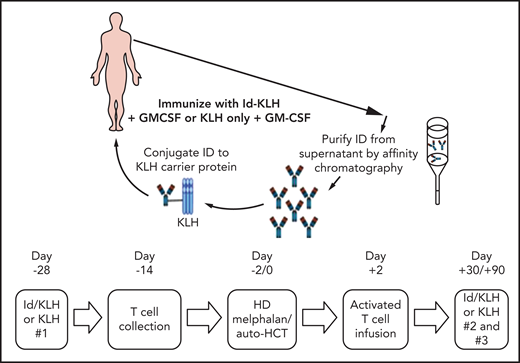
Generation of therapeutic T cells ex vivo
The apheresed mononuclear cells collected from patients were monocyte depleted via adherence to Dynal M-450 Epoxy beads or by counter flow centrifugal elutriation (Gambro Elutra Cell Separation System). The beads were then removed by magnetic separation by the MaxSep Magnetic Separator, and the number of recovered (monocyte-depleted) cells was determined. Cells were then grown in X-VIVO15 media supplemented with 5% commercial pooled human AB serum. Anti-CD3/anti-CD28 antibody–coated microbeads (Dynal; Thermo Fisher Scientific, Waltham, MA) were washed and added at a 3:1 ratio of beads per cell. The cultures were maintained for up to 12 days before harvesting and preparation for reinfusion. The target number of costimulated T cells for infusion was 1 × 1010 T cells total in 100 to 500 mL total volume. Manufacturing and release testing were performed in accordance with standard operating procedures. Final products were not released until all testing was performed and records were reviewed and signed off by quality assurance.
Outcomes and end points
Response and progression were defined according to the International Myeloma Working Group (IMWG) criteria.15 The severity of adverse events was assessed according to the Common Terminology Criteria for Adverse Events (CTCAE; version 3.0).16 High-risk chromosomal abnormalities were defined per the IMWG consensus panel.17 MM-specific patient assessments were performed at baseline, at 1 and 3 months after transplantation, and then every 3 months during the first year after auto-HCT. Additional laboratory tests and imaging studies were completed as clinically needed.
The primary efficacy end point was to evaluate whether infusions of Id-KLH–primed compared with non–Id-KLH CD3/CD28 activated autologous lymphocytes mediated a stronger treatment-related immune response. This was measured assessing differences in transcriptional profiles of CD8+/CD4+ T cells through the nCounter Gene Expression Profile assay (NanoString, Seattle, WA). Our initial immunological end points included evaluating the development of humoral immunity by detecting antibody titers using a panel of isotype-matched IDs from other patients as specificity controls. However, because of reagent qualification issues, low potency of Id-KLH, and high immunogenicity of KLH, an exogenous antigen, we could not adequately perform these humoral immunity assays. With this limitation, we elected to evaluate immunological responses by NanoString assays. The primary efficacy end point was evaluated at 6 months posttransplantation.
The primary safety end point was to evaluate the occurrence of treatment-related adverse events or treatment-related trial discontinuations defined as National Cancer Institute CTCAE grade ≥3 signs/symptoms, laboratory toxicities, and clinical events that were possible, likely, or definitely related to study treatment at any time from the first Id-KLH vaccine injection or the activated autologous T-cell infusion.
The secondary efficacy end point was complete response (CR)/very good partial response (VGPR) as defined by IMWG criteria. CR/VGPR were evaluated 90 days and toxicity was evaluated 30 days after autologous stem cell transplantation. If there were not at least 2 CRs/VGPRs among the first 14 patients or >2 patients with grade 4 to 5 nonhematological toxicities in the first 14 patients, the study arm was to be closed; otherwise, the accrual was to continue until the end of the study.
Statistical analysis
χ2 or Fisher’s exact test was used to examine differences of categorical variables, and Wilcoxon rank-sum or Kruskal-Wallis test was used to detect differences for continuous variables between groups.18 Progression-free survival (PFS) was defined as the time from transplantation to progression or death, whichever occurred first. Overall survival (OS) was defined as the duration from time of transplantation to the date of death. For events that had not occurred by the time of data analysis, times were censored at the last contact at which the patient was known to be progression free for PFS or alive for OS. Distributions of PFS and OS were estimated by the Kaplan-Meier method.19 Log-rank test20 was performed to test the difference in survival between groups. Regression analyses of survival data based on the Cox proportional hazards model21 were conducted on PFS. None of the covariates of interest violated the proportional hazards assumption of the Cox model. SAS (version 9.4) and S-Plus (version 8.04) were used to carry out the computations for all analyses.
NanoString analysis
CD4 and CD8 T-cell purification and isolation of mRNA
Single-cell suspensions of peripheral blood mononuclear cells (PBMCs) were subjected to magnetic bead separation. CD4+ or CD8+ T cells were isolated by the CD4+ or CD8+ Positive Selection Kit (Stemcell Technologies, Inc., Vancouver, Canada) after T-cell enrichment using the Pan T-cell Negative Selection Kit (Stemcell Technologies, Inc., Vancouver, Canada). Cell purity ≥95% was confirmed by analysis on the BD LSRFortessa flow cytometer after isolation. Data were analyzed using the FlowJo software package. Total RNA was extracted from isolated immune cells using the RNeasy Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Samples were eluted in 40 to 60 μL of RNase-free water and frozen at −80°C until analysis.
mRNA digital profiling
Gene expression was directly measured via counts of corresponding messenger RNA (mRNA) in each sample using the nCounter Human Immunology V2 Kit (NanoString, Seattle, WA), which is a multiplex assay for 594 genes involved in the human immunology response. Batches of 12 separate samples at 1 time were prepared as per the manufacturer’s instructions, with 100 to 300 ng of total RNA hybridized with probes at 65°C for 16 to 18 hours before being placed into the automated nCounter Prep Station (NanoString) in which samples were affixed to cartridges. Cartridges were then immediately placed into the nCounter Digital Analyzer (NanoString) optical scanner and read at a goal resolution of 550 fields of view, which is the maximum resolution for this instrument.
NanoString data preprocessing
The raw NanoString gene expression data were normalized using negative controls, positive controls, and housekeeping genes via nSolver (version 2.0) software (NanoString). The arithmetic means plus 2 standard deviations of the internal negative controls in each sample were subtracted from the gene expression count to ensure that any nonspecific mRNA detection was excluded. Values <0 were included as 0 for the analysis. The geometric mean of the 6 internal positive controls was used to normalize the data so that comparisons could be made across samples and to minimize distortion from batch effects.
Gene expression was then normalized to both spike-in positive control RNA and 6 housekeeping genes (ACTB, G6PD, OAZ1, POLR1B, POLR2A, RPL27, TPS13, and TBP) as previously described.22 Batch effects were assessed with a confirmatory mixed batch of samples selected from each prior run, with adjustments made as appropriate. The in-group means of gene expression count were compared via 2-tailed pairwise analysis with nonparametric distribution assumed. Given the number of simultaneous tests and an expected increase in type 1 errors, a Bonferroni-corrected threshold value of <.001 was used to indicate significance. Spearman correlation was used to cluster samples comparing overall expression levels. Logistic regression was used to compare the distribution of mean expression across the patients with or without clinical responses to this therapy.
Differential gene expression analysis
CD8+ and CD4+ T cells were isolated from PBMCs obtained pre- (T0) and at 3 time points postvaccine (days 30 [T30], 90 [T90], and 180 [T180]) after activated T-cell infusion) from 16 patients (8 patients treated with Id-KLH and 8 patients treated with KLH control). Gene expression profiling was batched, performed by NanoString, and fold change (FC) of each gene was calculated as the ratio of average gene expression intensity between pre- (T0) and postvaccine samples (T30, T90, and T180). P values were calculated from paired Student t tests between pre- (T0) and postvaccine (T30, T90, and T180). A gene was claimed to be differentially expressed if it showed an FC of >2 or −0.5 or less and P ≤ .05. Volcano plots were used to visualize log2 FC on the x-axis and paired P values from Student t tests between pre- (T0) and postvaccine (T30, T90, and T180) on the y-axis. Differentially expressed genes (P < .05 and FC >2 or −0.5 or less) between pre- and postvaccine were highlighted in red at different time points. The horizontal lines at y 1.31 represent the threshold of statistical significance (P = .05), and vertical lines at x = ±1 represent the threshold of 2 FCs set as cutoff values for the definition of differentially expressed genes.
Cytokine release by T cells stimulated with autologous Id protein–pulsed immortalized B cells
To establish autologous immortalized B cells to serve as antigen-presenting cells in vitro, we first introduced the genes encoding BCL-6 and BCL-XL into B cells, using retroviral vectors as previously described.23 For retroviral constructs and production of recombinant retrovirus, complementary DNA encoding BCL-6 and BCL-XL proteins was ligated into pRetro-X-IRES-ZsGreen1 vector (TaKaRa Bio USA, Mountain View, CA). The retroviral plasmids were transfected into the GP2-293 packaging cell line, which expresses gag and pol proteins (TaKaRa Bio USA). The envelope protein was supplied on a separate plasmid, phCMV-GALV-MTR (Addgene plasmid #163612), according to the manufacturer’s protocols. At 4 days after transfection, retroviral supernatant was collected and concentrated using Retro-X concentrator, and cell-free aliquots were stored at −80°C. Next, to generate autologous immortalized B cells, primary B cells were isolated from patient PBMCs using the EasySep Human B Cell Isolation Kit (#17954; Stemcell Technologies, Inc.). Isolated primary autologous B cells were activated by coculturing with irradiated (80 Gy) CD40L− mouse L cells and interleukin-21 (IL-21; 50 ng/mL) for 48 hours at 37°C before transduction. The activated primary autologous B cells were then ex vivo transduced with the BCL-6/BCL-XL recombinant retrovirus using the human fibronectin fragment CH-296 transduction protocol as described (RetroNectin; TaKaRa Bio USA).24 Transduction efficiency was measured by flow cytometry, and GFP+ CD19+ B cells were sorted out and freshly cultured with irradiated (80 Gy) CD40L− mouse L cells and IL-21 (50 ng/mL).
CD4+ T cells were isolated from either patient PBMCs or therapeutic activated T-cell products using the EasySep Human CD4+ T Cell Isolation Kit (#17952; Stemcell Technologies, Inc.). Isolated cells were expanded using a rapid expansion protocol (REP) as previously described.25 Briefly, the REP used OKT3 (anti-CD3) antibody (Ortho Biotech, Bridgewater, NJ) and IL-2 (100 IU/mL) in the presence of irradiated allogeneic feeder cells at a 200:1 ratio of feeder cells to patient CD4+ T cells. PBMC feeder cells were obtained from 3 normal volunteers by apheresis and were thawed, washed, and resuspended in 25 total mL of CTL media (RPMI 1640, GlutaMAX-HEPES, and 10% human AB serum; Thermo Fisher Scientific) and irradiated (50 Gy). PBMC feeder cells (1 × 108), OKT3 antibody (30 ng/mL), 25 mL of CTL media, and CD4+ T cells (0.5 × 106) were combined, mixed, and aliquoted to a 25-cm2 tissue culture flask. Flasks were incubated upright at 37°C in 5% CO2. Additional IL-2 was added at 100 IU/mL on day 2. On day 5, 20 mL of culture supernatant was removed by aspiration (cells were retained on the bottom of the flask), and media was replaced with a CTL media containing 100 IU/mL of IL-2. On day 6 and every day thereafter, the cell concentration was determined and cells were split into additional flasks or transferred to 75-cm2 tissue culture flasks with additional medium containing 100 IU/mL of IL-2 as needed to maintain cell densities at ∼1 × 106 cells per mL. Approximately 14 days after initiation of the REP, cells were harvested from the culture flask and were cryopreserved for future experimental analysis.
Irradiated washed autologous immortalized B cells were pulsed with patient-specific Id protein or KLH and were cocultured with REP CD4+ T cells in CTL media containing 10% human AB serum in 1 well of a 24-well plate. Cultures were restimulated with Id protein– or KLH-pulsed autologous immortalized B cells with IL-2 on day 11. After 2 separate stimulations 10 days apart (days 2 and 12), the CD4+ T cells were isolated on day 22. To test CD4+ T cell–specific reactivity to patient Id protein, all recognition assays were carried out in 96-well plates by coculture of 1 × 105 target cells (autologous immortalized B cells pulsed with patient Id [100 μg/mL], Id proteins from other patients [100 μg/mL] as negative controls for specificity, and KLH [100 μg/mL) as positive control protein) and 1 × 105 of the isolated stimulated T cells for 48 hours in 200 μL at 37°C in 5% CO2. The superantigen SEB (Staphylococcus aureus; enterotoxin type B) served as a positive control for CD4+ T-cell stimulation, and a no-antigen control group was also included. Supernatants were harvested, and GM-CSF and interferon-γ (data not shown) were assayed by enzyme-linked immunosorbent assay using coupled antibody pairs from R&D Systems (Minneapolis, MN).
Results
Patient characteristics
A total of 36 patients were enrolled between January 2013 and May 2015, 20 in the KLH-only arm and 16 in the Id-KLH arm, and completed their assigned treatments. The mean age of study participants was 59 years; 58% were men, and 78% were white. Twenty percent (7 of 36) of the cohort had high-risk cytogenetics, and additionally, 25% (9 of 36) had higher revised International Staging System stage 2 and 3 disease. A majority of patients (75%; 27 of 36) had achieved ≥PR to induction therapy before transplantation, with ≥VGPR rate of 14% (5 of 36). There were no significant differences between the 2 groups in age, sex, stage, cytogenetic risk, induction regimen, or response to induction (Table 1).
Patient characteristics
| Characteristic | Treatment arm | P | |
|---|---|---|---|
| KLH-only arm | Id-KLH arm | ||
| Sex | .82 | ||
| Female | 8 (40) | 7 (43.8) | |
| Male | 12 (60) | 9 (56.3) | |
| Race | .85 | ||
| Black | 2 (10) | 3 (18.8) | |
| M | 2 (10) | 1 (6.3) | |
| White | 16 (80) | 12 (75) | |
| Cytogenetic risk | .68 | ||
| High | 3 (15) | 4 (25) | |
| Standard | 17 (85) | 12 (75) | |
| International Staging System | .69 | ||
| I | 13 (72.2) | 7 (63.6) | |
| II, III | 5 (27.8) | 4 (36.4) | |
| LDH | 1.00 | ||
| >Normal | 2 (11.8) | 1 (8.3) | |
| Normal | 15 (88.2) | 11 (91.7) | |
| Response before transplantation | .35 | ||
| NCR | 0 (0) | 1 (6.3) | |
| VGPR | 1 (5) | 3 (18.8) | |
| PR | 14 (70) | 8 (50) | |
| SD | 2 (10) | 3 (18.8) | |
| PD | 3 (15) | 1 (6.3) | |
| Induction | .48 | ||
| VRD | 15 (75) | 10 (62) | |
| VCD | 4 (20) | 5 (31) | |
| Maintenance | 19 (95) | 16 (100) | .29 |
| Lenalidomide | 10 | 12 | |
| Lenalidomide based | 8 | 4 | |
| Pomalidomide | 1 | 0 | |
| Characteristic | Treatment arm | P | |
|---|---|---|---|
| KLH-only arm | Id-KLH arm | ||
| Sex | .82 | ||
| Female | 8 (40) | 7 (43.8) | |
| Male | 12 (60) | 9 (56.3) | |
| Race | .85 | ||
| Black | 2 (10) | 3 (18.8) | |
| M | 2 (10) | 1 (6.3) | |
| White | 16 (80) | 12 (75) | |
| Cytogenetic risk | .68 | ||
| High | 3 (15) | 4 (25) | |
| Standard | 17 (85) | 12 (75) | |
| International Staging System | .69 | ||
| I | 13 (72.2) | 7 (63.6) | |
| II, III | 5 (27.8) | 4 (36.4) | |
| LDH | 1.00 | ||
| >Normal | 2 (11.8) | 1 (8.3) | |
| Normal | 15 (88.2) | 11 (91.7) | |
| Response before transplantation | .35 | ||
| NCR | 0 (0) | 1 (6.3) | |
| VGPR | 1 (5) | 3 (18.8) | |
| PR | 14 (70) | 8 (50) | |
| SD | 2 (10) | 3 (18.8) | |
| PD | 3 (15) | 1 (6.3) | |
| Induction | .48 | ||
| VRD | 15 (75) | 10 (62) | |
| VCD | 4 (20) | 5 (31) | |
| Maintenance | 19 (95) | 16 (100) | .29 |
| Lenalidomide | 10 | 12 | |
| Lenalidomide based | 8 | 4 | |
| Pomalidomide | 1 | 0 | |
Data are presented as n (%).
LDH, lactate dehydrogenase; NCR, near CR; PD, progressive disease; SD, stable disease; VCD, bortezomib, cyclophosphamide, and dexamethasone; VRD, bortezomib, lenalidomide, and dexamethasone.
Toxicities
No infusion reactions or dose-limiting toxicities were seen in either arm. Most of the toxicities were limited to grade 1 or 2 (CTCAE; version 3.0). As outlined in Table 2, there were no significant differences in toxicities between the 2 arms except that more patients in the Id-KLH arm experienced grade 2 diarrhea than those in the KLH arm (31.3% vs 0%; P = .0306; Table 2). There were no treatment-related deaths in either arm at the 100-day and 1-year time points, with a nonrelapse mortality of 0. Nineteen (95%) and 16 patients (100%) received maintenance therapy after protocol treatment in the KLH-only and Id-KLH arms, respectively (P = 1.0). Patients continued maintenance therapy until disease progression or until it resulted in unacceptable toxicity.
Toxicities by treatment arm
| Grade | Treatment arm | P | |
|---|---|---|---|
| KLH-only arm | Id-KLH arm | ||
| Diarrhea | .0306 | ||
| No toxicities | 10 (50) | 6 (37.5) | |
| 1 | 10 (50) | 5 (31.3) | |
| 2 | 0 (0) | 5 (31.3) | |
| Dysphagia/mucositis | 1.000 | ||
| No toxicities | 15 (75) | 12 (75) | |
| 1 | 5 (25) | 4 (25) | |
| Nausea | .1756 | ||
| No toxicities | 7 (35) | 4 (25) | |
| 1 | 5 (25) | 1 (6.3) | |
| 2 | 7 (35) | 11 (68.8) | |
| 3 | 1 (5) | 0 (0) | |
| Transaminase elevation | |||
| No toxicities | 20 (100) | 16 (100) | |
| Bilirubin elevation | .3671 | ||
| No toxicities | 17 (85) | 12 (75) | |
| 1 | 3 (15) | 2 (12.5) | |
| 2 | 0 (0) | 2 (12.5) | |
| Grade | Treatment arm | P | |
|---|---|---|---|
| KLH-only arm | Id-KLH arm | ||
| Diarrhea | .0306 | ||
| No toxicities | 10 (50) | 6 (37.5) | |
| 1 | 10 (50) | 5 (31.3) | |
| 2 | 0 (0) | 5 (31.3) | |
| Dysphagia/mucositis | 1.000 | ||
| No toxicities | 15 (75) | 12 (75) | |
| 1 | 5 (25) | 4 (25) | |
| Nausea | .1756 | ||
| No toxicities | 7 (35) | 4 (25) | |
| 1 | 5 (25) | 1 (6.3) | |
| 2 | 7 (35) | 11 (68.8) | |
| 3 | 1 (5) | 0 (0) | |
| Transaminase elevation | |||
| No toxicities | 20 (100) | 16 (100) | |
| Bilirubin elevation | .3671 | ||
| No toxicities | 17 (85) | 12 (75) | |
| 1 | 3 (15) | 2 (12.5) | |
| 2 | 0 (0) | 2 (12.5) | |
Data are presented as n (%).
Clinical outcomes
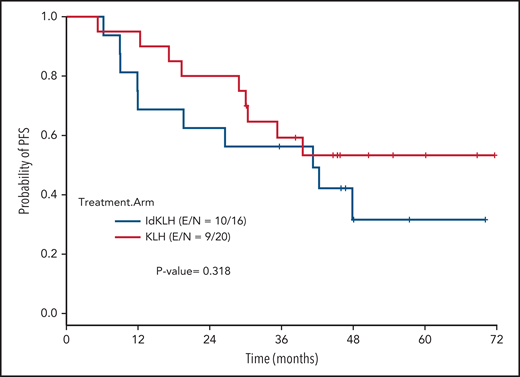
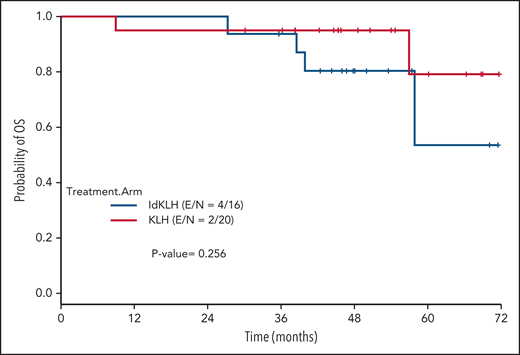
At last evaluation, 6 (30%) and 8 patients (50%) had achieved CR in the KLH-only and Id-KLH arms, respectively (P = .22), and 13 (65%) and 12 (75%) had achieved at least VGPR in the KLH-only and Id-KLH arms, respectively (0.72). The median follow-up of the entire cohort was 48.6 months (range, 9.0-71.7). There were no differences in PFS between the 2 arms (P = .32), with 3-year PFS rates of 59.2% (95% confidence interval [CI], 41.0%-85.7%) and 56.3% (95% CI, 36.5%-86.7%) in the KLH-only and Id-KLH groups, respectively (Figure 2). At the time of last follow-up, 6 of 36 patients had died, all as a result of recurrence of disease, 2 in the KLH-only arm and 4 in the Id-KLH arm, respectively. The 3-year OS rates in the KLH and Id-KLH groups were 95% (95% CI, 85.9%-100%) and 93.8% (95% CI, 82.6%-100%), respectively (Figure 3).
Immune response assessment by gene expression
Experimental treatment-related immune responses were evaluated by measuring the differences in transcriptional profiles of CD8+/CD4+ T cells between the KLH-only and Id-KLH arms. Eight and 3 patients in each arm had sufficient T cells available at the pre- (T0) and postvaccine time points (T30, T90, and T180), respectively, for complete analysis. A total of 31, 27, and 20 genes were identified as differentially expressed (FC absolute value >2; P < .05) at the T30, T90, and T180 time points, respectively, compared with T0 prevaccine samples (Figure 4A). Changes in expression of these genes were observed almost exclusively in patients who had received the Id-KLH vaccine (Figure 4B).
![Differential CD8+ T-cell immune gene expression between patients receiving vaccine and control vaccine and responders (Rs) vs nonresponders (NRs). (A) CD8+ T cells were isolated from PBMCs obtained pre- (T0) and at 3 time points postvaccine (30 [T30], 90 [T90], and 180 [T180] days after activated T-cell infusion) from 16 patients (8 patients treated with Id-KLH and 8 patients treated with KLH control). Gene expression profiling was batched and performed by NanoString, and postvaccine results for each patient were compared against corresponding T0 prevaccine samples. Volcano plots illustrate the log2 FC in gene expression (x-axis) and paired P values from Student t tests between prevaccine (T0) and Id-KLH postvaccine samples (T30, T90, and T180; y-axis). Differentially expressed genes (P < .05 and log2 FC >2 or −0.5 or less) are indicated in red. (B) Heatmap displaying expression of 50 differentially expressed genes, as defined in panel A. Pooled results for all patients are shown for each gene in the column to the far right for each treatment arm. Pooled results are classified as significantly upregulated (red), significantly downregulated (blue), or no significant change (gray). (C) Volcano plot showing the distribution of FCs in gene expression comparing R and NR CD4+ T cells obtained at day +180 postvaccination. Genes with absolute FC >1.5 and adjusted P value false-discovery rate (FDR) <0.05 are indicated in red. (D) Serial expression of 36 differentially upregulated genes expressed between Rs and NRs across T0, T30, T90, and T180 time points. Mean T-cell mRNA expression level for each gene is shown. Differences in the mRNA copy numbers between time points were analyzed using a 2-tailed paired Student t test. Expression levels of the PYCARD gene are indicated in red.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/9/10.1182_blood.2020008493/4/m_bloodbld2020008493f4.png?Expires=1770432677&Signature=h-TxiqmhCviaCalvG3ra8~KSpTVqGuY3wZQfvKdEGyKO-8XJhInGfYdYsrpgMtEpQPr1WmcMHktW5GFMFID9AavXCpZ9xSkStdgjR6DSQqRtOFZysc-DoaqLf4IuVkqGeR6UYaf6s6LSxakD6zjmjA01ElIxQ6Sl0NKXM~g33EdgqYSvUyl4VQjku2j98u8GkNJsdl15edFLi0Sf344C-0KtJC4X1jNaqpLwBAf~qoxL6lKC-GOSXGnyK~4ala3u6NT4-Ou7-tGbvUsAJ0HHe60nAuAETy7mgXTvzzEeU0RybleWzzPGCOOepWqdtOxt53PQA8kLjw8CmhV063d1-g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Differential CD8+ T-cell immune gene expression between patients receiving vaccine and control vaccine and responders (Rs) vs nonresponders (NRs). (A) CD8+ T cells were isolated from PBMCs obtained pre- (T0) and at 3 time points postvaccine (30 [T30], 90 [T90], and 180 [T180] days after activated T-cell infusion) from 16 patients (8 patients treated with Id-KLH and 8 patients treated with KLH control). Gene expression profiling was batched and performed by NanoString, and postvaccine results for each patient were compared against corresponding T0 prevaccine samples. Volcano plots illustrate the log2 FC in gene expression (x-axis) and paired P values from Student t tests between prevaccine (T0) and Id-KLH postvaccine samples (T30, T90, and T180; y-axis). Differentially expressed genes (P < .05 and log2 FC >2 or −0.5 or less) are indicated in red. (B) Heatmap displaying expression of 50 differentially expressed genes, as defined in panel A. Pooled results for all patients are shown for each gene in the column to the far right for each treatment arm. Pooled results are classified as significantly upregulated (red), significantly downregulated (blue), or no significant change (gray). (C) Volcano plot showing the distribution of FCs in gene expression comparing R and NR CD4+ T cells obtained at day +180 postvaccination. Genes with absolute FC >1.5 and adjusted P value false-discovery rate (FDR) <0.05 are indicated in red. (D) Serial expression of 36 differentially upregulated genes expressed between Rs and NRs across T0, T30, T90, and T180 time points. Mean T-cell mRNA expression level for each gene is shown. Differences in the mRNA copy numbers between time points were analyzed using a 2-tailed paired Student t test. Expression levels of the PYCARD gene are indicated in red.
Differential CD8+ T-cell immune gene expression between patients receiving vaccine and control vaccine and responders (Rs) vs nonresponders (NRs). (A) CD8+ T cells were isolated from PBMCs obtained pre- (T0) and at 3 time points postvaccine (30 [T30], 90 [T90], and 180 [T180] days after activated T-cell infusion) from 16 patients (8 patients treated with Id-KLH and 8 patients treated with KLH control). Gene expression profiling was batched and performed by NanoString, and postvaccine results for each patient were compared against corresponding T0 prevaccine samples. Volcano plots illustrate the log2 FC in gene expression (x-axis) and paired P values from Student t tests between prevaccine (T0) and Id-KLH postvaccine samples (T30, T90, and T180; y-axis). Differentially expressed genes (P < .05 and log2 FC >2 or −0.5 or less) are indicated in red. (B) Heatmap displaying expression of 50 differentially expressed genes, as defined in panel A. Pooled results for all patients are shown for each gene in the column to the far right for each treatment arm. Pooled results are classified as significantly upregulated (red), significantly downregulated (blue), or no significant change (gray). (C) Volcano plot showing the distribution of FCs in gene expression comparing R and NR CD4+ T cells obtained at day +180 postvaccination. Genes with absolute FC >1.5 and adjusted P value false-discovery rate (FDR) <0.05 are indicated in red. (D) Serial expression of 36 differentially upregulated genes expressed between Rs and NRs across T0, T30, T90, and T180 time points. Mean T-cell mRNA expression level for each gene is shown. Differences in the mRNA copy numbers between time points were analyzed using a 2-tailed paired Student t test. Expression levels of the PYCARD gene are indicated in red.
CD8+ T cells
Analysis of individual differentially expressed genes (Figure 4B) revealed that 6 genes were significantly upregulated at multiple postvaccine time points, including CCL5, which is highly expressed in memory T cells.26 In addition, granzyme-A (GZMA) and granulysin (GNLY), which are indicators of cytotoxic T-cell effector function, were significantly increased, as was the transcription factor TBX21 (encoding T-bet), which is thought to play a pivotal role in CD8+ T-cell differentiation26 and in maintenance of memory subpopulations, as well as in clearance of acute viral infections.27 Finally, genes associated with an early effector memory and terminal effector memory phenotype,28 including B3GAT1 (β-1,3-glucuronyltransferase-1; CD57), KLRD1, and KLRG1, were significantly overexpressed.
Among patients who had received the Id-KLH vaccine, several genes were downregulated postvaccination, most with unclear function. However, several genes with known negative regulatory function in T cells were among the downregulated genes, including PTK2,29LILRB3,30TGB1,31 and LGALS3.32-34 Five other genes potentially involved in CD8+ T-cell activation (CSF2RB [CD131], CSF3R [CD114], TNFRSF10C [TRAIL-R3; DCR1], NFKBIZ [IκB], and ZBTB16 [PLZF]) were consistently downregulated post-vaccination, which is of unclear significance.
CD4+ T cells
We also performed gene expression profiling of CD4+ T cells. A total of 30 genes were differentially expressed between pre- (T0) and any of the 3 postvaccine time points among patients receiving Id-KLH. Among these, TBX21 gene expression consistently increased, whereas mRNA encoding for SOCS3 decreased across all 3 postvaccination time points (supplemental Figure 1). Elevated TBX21 expression is required for maximal clonal expansion and for the formation of terminal differentiation of CD4+ T effector cells.35SOCS3 is a negative regulator of T-cell function and is involved in the exhaustion of T cells.36
In contrast, in the KLH group, there were fewer genes consistently differentially expressed across multiple time points postvaccination, when compared with results from the Id-KLH group. In particular, genes associated with CD8+ T-cell effector function, activation, costimulation, or regulation were not significantly up- or downregulated (supplemental Figure 2). Among CD4+ T cells, 4 genes, CR1, MUC1, NT5E, and TNFRSF11A, were consistently elevated postvaccination. CR1,37NT5E, and MUC138 are negative regulators of CD4+ T-cell activity (supplemental Figure 3). Taken together, these results suggest successful activation, induction of effector function, and generation of memory CD8+ T cells after Id-KLH but not after KLH control vaccination.
Because our study did not show a difference between the arms in clinical outcomes, we examined patterns of immune response gene expression by comparing patients achieving a CR or VGPR (Rs; n = 13) and all other patients (NRs; n = 3) across both arms. At the day +180 time point, differential gene expression analysis of CD4+ T cells identified 65 differentially expressed genes, of which 36 were upregulated and 29 were downregulated (FC absolute vale >1.5; adjusted P value false-discovery rate <0.05; Figure 4C; supplemental Tables 1 and 2). Interestingly, upregulated genes included several well-known genes related to T-cell receptor (TCR) signal activation, such as SLAMF6,39CIHS,40 and BCAP31,41 which induce TCR stimulation. Among differentially downregulated genes, LGALS342 and CD543,44 are known to be negative
Several other distinct differentially upregulated genes included human effector memory T-cell markers KLRG145 (FC = 1.9; P = .033), KLRF146 (FC = 3.58; P = .025), and CD8647 (FC = 2.31; P = .035), as well as CCR548,49 (FC = 4.41; P = .022), which has been shown to boost antitumor responses. Most notably, there was strikingly significant upregulation of PYCARD (FC = 4.69; P = .0001). In mouse models, PYCARD−/− T regulatory cells exhibit greater suppressive capacity,50 suggesting that T regulatory cells may have been more activated in NR patients. Furthermore, the mRNA expression levels of these 36 differentially upregulated genes generally increased over time between T0 and T180 in R patients, while generally decreasing in NR patients, especially for PYCARD, consistent with successful T-cell activation associated with Id-KLH vaccination (Figure 4D). These findings may also guide future correlative analyses between immunological and clinical outcomes of this vaccine. Differential gene expression was not observed for CD8+ T cells.
Immune response assessment by functional T-cell cytokine secretion
We analyzed the achievement of myeloma Id–specific immunity by determining the ability of T cells from patients in the Id-KLH group to produce cytokines after vaccination (Figure 5A). Sufficient postvaccination PBMC samples (day +180) were available from 5 patients who had received Id-KLH vaccines for this functional analysis. Three of these 5 patients demonstrated specific CD4+ T-cell immune responses, as demonstrated by significantly increased levels of GM-CSF secretion in response to autologous antigen-presenting cells pulsed with autologous Id protein, compared with either no antigen or Id proteins from other patients (patients 256, 255, and 263; Figure 5B). T cells from these patients also responded to KLH as an internal control antigen. Stimulation with SEB was also included as another positive control (data not shown). Similarly, T cells isolated from cryopreserved therapeutic activated T-cell products from 3 patients receiving Id-KLH were tested for antigen-specific responses (the number of T-cell products available was limited by availability of corresponding autologous PBMCs required as antigen-presenting cells). All 3 T-cell products also demonstrated myeloma Id–specific secretion of GM-CSF (Figure 5C). Taken together, these data suggest functional correlation between Id-specific T cells and global changes in T-cell gene expression.
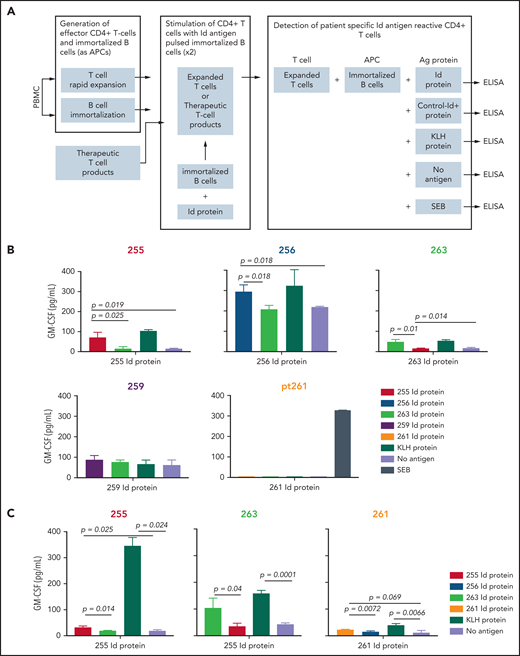
Detection of Id antigen–specific CD4+ T cells in postvaccination PBMCs (day +180). (A) Schematic showing the experimental design for detection of antigen-specific T-cell responses after 2 rounds of stimulation with autologous protein antigen-loaded immortalized B cells as antigen-presenting cells, as described in “Methods.” Mean GM-CSF concentration in culture supernatants after coculture of Id protein– or KLH (internal control)–loaded immortalized autologous B cells (or SEB; data not shown) by in vitro–expanded peripheral blood CD4+ T cells (B) or therapeutic activated T-cell products (C). Numbers refer to individual patients who had received Id-KLH immunization. Corresponding Id proteins from patients 263 and 255 served as specificity controls for other patients. Differences in cytokine concentrations were analyzed using a 2-tailed unpaired Student t test. ELISA, enzyme-linked immunosorbent assay.
Detection of Id antigen–specific CD4+ T cells in postvaccination PBMCs (day +180). (A) Schematic showing the experimental design for detection of antigen-specific T-cell responses after 2 rounds of stimulation with autologous protein antigen-loaded immortalized B cells as antigen-presenting cells, as described in “Methods.” Mean GM-CSF concentration in culture supernatants after coculture of Id protein– or KLH (internal control)–loaded immortalized autologous B cells (or SEB; data not shown) by in vitro–expanded peripheral blood CD4+ T cells (B) or therapeutic activated T-cell products (C). Numbers refer to individual patients who had received Id-KLH immunization. Corresponding Id proteins from patients 263 and 255 served as specificity controls for other patients. Differences in cytokine concentrations were analyzed using a 2-tailed unpaired Student t test. ELISA, enzyme-linked immunosorbent assay.
Analysis of T-cell exhaustion in Id-KLH and KLH-only arms
We also investigated the possibility of underlying immune suppression in MM as a barrier to effective vaccination. To this end, we characterized the expression of circulating T-cell exhaustion markers at the T1 time point (before vaccination) and compared it with that of healthy donors (n = 3) by RNA expression. Among CD8+ T cells, there was a general trend toward increased expression of exhaustion markers in the KLH control group (n = 8; 3 clinical Rs and 5 NRs), and these differences reached statistical significance, despite small sample sizes, for the LAG3, CD244, and KLRG1 genes, compared with healthy donors (supplemental Figure 4). Similarly, a trend toward increased exhaustion marker expression was observed in patients receiving Id-KLH (n = 7; all clinical Rs), but these differences were not significant. No significant differences in exhaustion marker expression were observed among CD4+ T cells in either the Id-KLH or KLH-only group. A majority of patients with MM in this study demonstrated significantly elevated RNA expression levels of CD8+ T-cell exhaustion markers at baseline.
Discussion
In this randomized trial, we used a prime-and-boost strategy by collecting T lymphocytes from patients that had been vaccine primed in vivo with patient-specific Id-KLH or KLH-only vaccine, activating and expanding them ex vivo with CD3/CD28 magnetic beads and reinfusing these cells after high-dose chemotherapy and auto-HCT. We showed that Id-KLH vaccination and vaccine-primed costimulated T cells could be safely administered in the setting of auto-HCT. We also showed a more robust immune response in CD4+ and CD8+ T cells and generation of vaccine-specific immunity in the Id-KLH arm.
This study showed that Id-KLH–primed costimulated T cells can be administered in the setting of auto-HCT without significant adverse reactions. There were no infusion reactions in either arm with the costimulated T cells, and no significant differences in toxicity between the 2 arms, except for an increase in diarrhea in the Id-KLH arm. The reason for diarrhea is not clear, although it was probably related to high-dose melphalan. Most diarrheal episodes resolved either spontaneously or with minimal treatment. We did not see any cytokine release syndrome, neurotoxicity, or graft-versus-host disease in either arm.
In our trial, patients receiving the Id-KLH vaccination showed a similar depth of response, with no significant differences in PFS or OS between the 2 arms. These outcomes were consistent with other trials using tumor vaccine–primed adoptive T cells in MM, where systemic vaccine-specific immunological responses did not translate into clinical efficacy.4,11-13,51 Rapaport et al13 performed a phase 1/2 trial to evaluate in vivo pneumococcal vaccine–primed and ex vivo–stimulated autologous T cells at different time points post–auto-HCT followed by booster doses of pneumococcal vaccine immunizations in patients with MM. They reported that autologous T-cell infusion closer to high-dose chemotherapy and stem cell infusion on day 12, compared with day 42 or 100, was more effective in reconstitution of T-cell immunity. In a subsequent trial,11 T cells were in vivo primed using a multipeptide antimyeloma tumor antigen derived from human telomerase reverse transcriptase and survivin. These T cells were infused at an earlier time point at day 2 post–auto-HCT. With this strategy, there was an augmented and accelerated cellular and humoral immune reconstitution, including antitumor immunity. However, as in our study, there was no difference in event-free survival or OS between the 2 arms in both these trials. The similar PFS in both arms despite a more robust antitumor immune response in the Id-KLH arm was probably due to small sample size and the unfavorable immune microenvironment in MM.
The primary hypothesis of our study was that the combination of Id-KLH vaccination and Id-KLH–primed T cells would induce a more robust antimyeloma immune response than the nonspecific KLH vaccine and KLH-primed T cells. Analysis of differentially expressed immune response genes showed higher activation, induction of effector function, and generation of memory CD8+ T cells after Id-KLH but not after the KLH control vaccination. The upregulation of these associated genes was persistent and could be seen up to day +180 post–auto-HCT. Differentially upregulated genes associated with T-cell activation were also observed in patients who achieved a CR or VGPR across both study arms, which may guide future correlative analyses of this and other vaccine therapies. These changes in T-cell gene expression were associated with functional myeloma Id–specific immune responses in the blood and therapeutic T-cell products of a subset of patients who had received Id-KLH vaccination.
Another important finding was significantly elevated RNA levels of CD8+ T-cell exhaustion markers in a majority of patients with MM at baseline, highlighting an immunosuppressed phenotype in treated patients with MM, as reported by other groups.52,53 Underlying immune suppression is a well-recognized barrier to effective active immunotherapy strategies.54 Recent reports suggest that cancer vaccines combined with checkpoint inhibitors may induce stronger antitumor immune responses, which may translate into better clinical responses and disease control.55 Future studies should focus on combining active immunization with agents such as checkpoint inhibitors or other immunomodulatory therapies that can overcome preexisting immunosuppression.55
To augment the antimyeloma immune responses, other vaccine strategies, such as the use of antigen-presenting dendritic cells primed with tumor lysates/peptides56 or fusion with tumor cells,57 have been employed, with promising initial results that have led to ongoing phase 2 trials. However, 2 decades of research with limited success have dampened the enthusiasm for vaccine-based approaches in antimyeloma therapy. With the advent of chimeric antigen receptor (CAR) T cell–based therapy and its excellent efficacy,58 we anticipate that the antimyeloma immune-based therapy will move away from vaccine-based approaches to CAR T cells and other cellular therapy approaches. Another area of investigation would be to use Id-KLH–primed T cells, as opposed to unprimed T cells, for autologous CAR T-cell generation, where the endogenous TCR is retained.
In summary, in this randomized phase 2 trial, we showed that Id-KLH vaccine–specific adoptive T cells can be safely administered in the setting of auto-HCT and induce a robust T-cell response. Combining this treatment with a checkpoint inhibitor or other immunomodulatory therapies may further improve the efficacy of this approach.
Authorship
Contribution: M.H.Q. designed and supervised the study, interpreted the data, and wrote the manuscript. N.Y.S. interpreted the data and wrote the first version of the manuscript. S.-c.C., Z.W., S.S.R., S.S., T.Z., and A.A. completed experiments and collected results of the study and critically analyzed the manuscript. H.L. and V.B. performed statistical analysis and critically analyzed the manuscript. E.A.S., B.T., M.H., K.K., M.P., Q.B., E.J.S., R.Z.O., B.L.L., N.K., A.G., A.C., D.T.V., K.D., C.H.J., and R.C. critically discussed the manuscript. L.W.K. designed and supervised the study, analyzed the data, and revised the manuscript.
Conflict-of-interest disclosure: M.H.Q. declares research funding from Janssen, Bioline, Angiocrine, Amgen, and NexImmune and has served on advisory boards for Bristol-Myers Squibb and Oncopeptides. R.Z.O. declares laboratory research funding from BioTheryX and clinical research funding from CARsgen Therapeutics, Celgene, Exelixis, Janssen Biotech, Sanofi-Aventis, and Takeda Pharmaceuticals North America, Inc.; has served on advisory boards for Amgen, Bristol-Myers Squibb, Celgene, EcoR1 Capital LLC, Forma Therapeutics, Genzyme, GSK Biologicals, Ionis Pharmaceuticals, Inc., Janssen Biotech, Juno Therapeutics, Kite Pharma, Legend Biotech USA, Molecular Partners, Regeneron Pharmaceuticals, Inc., Sanofi-Aventis, Servier, and Takeda Pharmaceuticals North America, Inc.; has served as a consultant for STATinMED Research; and is a founder of Asylia Therapeutics, Inc., with associated patents and an equity interest, although this technology does not bear on the current manuscript. B.L.L. and C.H.J. are scientific founders of Tmunity and have equity in Tmunity Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Muzaffar H. Qazilbash, Department of Stem Cell Transplantation and Cellular Therapy, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Houston, TX 77030; e-mail: mqazilba@mdanderson.org.
Presented in abstract form at the 59th Annual Meeting of the American Society of Hematology, Atlanta, GA, 9-12 December 2017.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal