

Key Points
Preclinical mouse modeling of cGVHD demonstrates persistent alterations in behavior.
CNS cGVHD presents with immunological features distinct from CNS acute GVHD, indicative of divergent mechanisms.

Abstract
Graft-versus-host disease (GVHD) remains the leading cause of nonrelapse mortality after allogeneic stem cell transplantation for hematological malignancies. Manifestations of GVHD in the central nervous system (CNS) present as neurocognitive dysfunction in up to 60% of patients; however, the mechanisms driving chronic GVHD (cGVHD) in the CNS are yet to be elucidated. Our studies of murine cGVHD revealed behavioral deficits associated with broad neuroinflammation and persistent Ifng upregulation. By flow cytometry, we observed a proportional shift in the donor-derived T-cell population in the cGVHD brain from early CD8 dominance to later CD4 sequestration. RNA sequencing of the hippocampus identified perturbations to structural and functional synapse-related gene expression, together with the upregulation of genes associated with interferon-γ responses and antigen presentation. Neuroinflammation in the cortex of mice and humans during acute GVHD was recently shown to be mediated by resident microglia-derived tumor necrosis factor. In contrast, infiltration of proinflammatory major histocompatibility complex (MHC) class II+ donor bone marrow (BM)–derived macrophages (BMDMs) was identified as a distinguishing feature of CNS cGVHD. Donor BMDMs, which composed up to 50% of the CNS myeloid population, exhibited a transcriptional signature distinct from resident microglia. Recipients of MHC class II knockout BM grafts exhibited attenuated neuroinflammation and behavior comparable to controls, suggestive of a critical role of donor BMDM MHC class II expression in CNS cGVHD. Our identification of disease mediators distinct from those in the acute phase indicates the necessity to pursue alternative therapeutic targets for late-stage neurological manifestations.
Introduction
Chronic graft-versus-host disease (cGVHD) is the leading cause of nonrelapse mortality after otherwise curative allogeneic stem cell transplantation (SCT) for hematological malignancies.1,2 cGVHD can be inflammatory and/or fibrotic with organ-specific or multiorgan symptomatology, and complex and protean presentations make diagnosis challenging.3 Manifestations of cGVHD in the central nervous system (CNS) have recently been recognized as a clinical entity independent of complications associated with pretransplantation conditioning and immunotherapy.4 Up to 60% of adult SCT survivors experience neurocognitive dysfunction, evidenced by impaired learning and memory, concentration deficits, and emotional changes, including anxiety and depression,5 with reports of underlying cerebral vascular pathologies and encephalitis.4 Although recent studies have begun to elucidate the pathophysiology of CNS acute GVHD (aGVHD),6,7 we report here the first preclinical evidence of CNS cGVHD. GVHD is considered the primary determinant of after transplant quality of life8; therefore, understanding the effects of complex immunopathology on brain function serves to identify targets for potential preventative and therapeutic treatments.
Consistent with alloreactive donor T cells driving peripheral GVHD pathology,9,10 murine and nonhuman primate CNS aGVHD models showed that alloreactive T cells infiltrate the CNS and induce neuronal damage.6,7,11 Postmortem brain samples and experimental disease models, such as in Alzheimer’s disease and multiple sclerosis/experimental autoimmune encephalitis, suggest that CNS infiltration of pathogenic T cells with associated proinflammatory cytokine production is a common feature of chronic neuroinflammation.12,13 CNS damage and disease typically involve activation of tissue-resident microglia. As long-lived macrophages, microglia conduct dynamic immune surveillance, among other functions, including provision of trophic support and regulation of synaptic plasticity.14,15 Inflammatory stimuli induce a reactive phenotype characterized by morphological changes and cytokine release. Microglia may restore a quiescent state after activation or contribute to ongoing inflammation,16 exemplified by the recent description of microglial tumor necrosis factor (TNF) production as a critical mediator of CNS aGVHD.7 Additionally, disease models demonstrate a detrimental contribution of CNS-infiltrating bone marrow (BM)–derived macrophages (BMDMs),17 similar to cutaneous cGVHD induction by infiltrating donor-derived macrophages.18 Although CNS-infiltrating macrophages adopt microglial morphology and identifying surface marker (eg, ionized calcium binding adaptor molecule-1 [Iba1]) expression, widespread profiling suggests a functionally and transcriptionally distinct population.19,20 Elucidating resident microglia phenotype and function and the temporal contribution of donor BMDMs will be instrumental for understanding biological disease mechanisms and informing effective therapeutic strategies late after transplant.
The necessity for a controlled immunological environment to maintain optimal brain function underlies the hypothesis that GVHD-induced inflammatory changes are modulating behaviors. Here, we aimed to characterize CNS cGVHD and provide the first evidence of prolonged behavioral deficits dependent on sustained inflammation. Overall, we confirm the brain as a novel cGVHD target organ where neuroinflammation is characterized by dysregulated cytokine production and infiltration donor-derived major histocompatibility complex (MHC) II–expressing macrophages.
Materials and methods
Mice
Female mice between ages 8 and 12 weeks were used, housed in sterilized microisolator cages with acidified autoclaved water (pH, 2.5) and food available ad libitum. Strains used are listed in supplemental Table 1. Experiments were approved by and performed in accordance with the QIMR Berghofer Animal Ethics Committee (Brisbane, Queensland, Australia).
BM transplantation
On day 0, recipients underwent 1100 (B6D2F1) or 1000 cGy (C57BL/6) split-dose total-body irradiation (casesium-137 source) and received 5 × 106 (B6D2F1) or 10 × 106 (C57BL/6) T cell–depleted (TCD) BM alone (non-GVHD controls) or with 0.5 × 106 C57BL/6 (B6D2F1) or 5 × 106 BALB/c (C57BL/6) splenic T cells enriched by BioMag (Qiagen) bead depletion of non-T cells to induce GVHD.21 MHC II knockout (KO) donor grafts contained TCD BM only. Mice were monitored daily to evaluate clinical GVHD scores as previously published.22
Behavioral testing
Flow cytometry and cell sorting
Surface staining of single cell suspensions of digested brain was performed in fluorescence-activated cell sorting (FACS) buffer (phosphate buffered saline, 2% fetal calf serum, and 5 mM of EDTA) at room temperature for 15 minutes in darkness. The antibodies used are listed in supplemental Table 2. Flow cytometric acquisition was performed with an LSRFortessa cytometer (BD Biosciences), and data analyzed using FlowJo software (version 10). Cell sorting was performed using an FACSAria III Cell Sorter (BD Biosciences).
Statistics
GraphPad Prism software (version 7.0) was used to conduct all statistical analyses. An unpaired 2-tailed Student t test was used for comparison of 2 groups, and a 1-way analysis of variance (ANOVA) with Tukey multiple comparisons was used for GVHD groups across 3 time points. Longitudinal behavior data were analyzed with repeated measures 2-way ANOVA with Bonferroni post hoc comparison, and clinical scores were analyzed with a 2-way ANOVA with Bonferroni multiple comparisons test. All data are presented as mean ± standard error of the mean, with significance at P < .05.
Details of behavioral tests, devices used, and standard procedures including tissue procurement and processing, flow cytometry, immunofluorescence and imaging acquisition, cell quantification, quantitative real-time polymerase chain reaction (oligonucleotide sequences listed in supplemental Table 3), and RNA sequencing (RNA-seq) are provided in the data supplement.
Results
cGVHD induces persistent behavioral deficits
We used established MHC-mismatched cGVHD models10 (Figure 1A-B), where transfer of a low T-cell dose induces nonlethal disease with subsequent development of sclerodermatous skin pathology after transplant (supplemental Figure 1A-D). Preceded by low-grade aGVHD, cGVHD develops in both models by day 35, with persistent pathology evident in the later stages of disease (days 70-100). To measure neurological manifestations of cGVHD, we first used a battery of behavioral tests in the B6 into B6D2F1 model. cGVHD mice showed normal grip strength (supplemental Figure 1E) and exploratory behavior (supplemental Figure 1F) in the rotarod and open field tests, respectively, with no evidence of anxiety-like behavior in the elevated plus maze (supplemental Figure 1G). In support of recent findings,7 GVHD mice demonstrated poor recognition memory in the novel object recognition test at day 14; however, this did not persist into the chronic stages of disease (supplemental Figure 1H). However, using the FST to assess the behavioral response of cGVHD mice to an aversive situation,29 we confirmed in our model the previous report that aGVHD mice (day 14) exhibit increased mobility as a dysfunctional response in the FST30 (supplemental Figure 1I). Moreover, testing at both early and late phases of cGVHD (days 35 and 70, respectively) demonstrated significantly increased mobility in cGVHD mice compared with TCD controls (Figure 1C). Extending these findings, we confirmed dysfunctional responses in the FST during the acute phase (day 14; supplemental Figure 1J) and at both early- and late-stage cGVHD using the BALB/c into B6 model (Figure 1D). We next used the active place avoidance paradigm to assess spatial learning and memory, which have been extensively shown to depend on the integrity of the hippocampus.31,32 This task requires mice to integrate visuospatial room cues to avoid a concealed shock zone on a rotating platform.24 We first confirmed that TCD mice effectively completed this task in a manner similar to naïve age-matched controls, thus indicating intact working spatial memory (supplemental Figure 1K). Compared with TCD controls, however, cGVHD mice exhibited learning deficits evidenced by increased entries into the shock zone and less time spent avoiding the shock zone (Figure 1E-F). Improvement over the 5-day testing period beginning at day 70 (Figure 1F) was significantly reduced in cGVHD mice compared with TCD controls. Conclusively, these novel data indicate that cGVHD mice exhibit pronounced alterations in behavior persisting late after transplant.
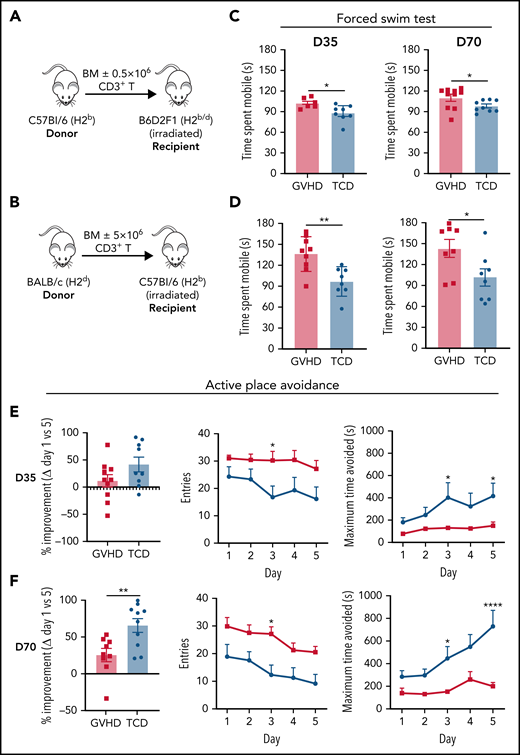
cGVHD induces prolonged behavioral deficits. (A) Schematic of transplantation regime. Lethally irradiated B6D2F1 (H2b/d) recipient mice received 5 × 106 TCD BM with no T cells or with 0.5 × 106 CD3+ T cells from C57Bl/6 (H2b) donors to induce low-grade nonlethal cGVHD. (B) Schematic of transplantation regime. Ten × 106 BM with or without 5 × 106 CD3+ T cells from a BALB/c (H2d) donor were transplanted into lethally irradiated C57Bl6 (H2b) recipients. (C) Time B6D2F1 recipients spent swimming (mobile) in the forced swim test at days 35 and 70 after transplant (day 35: n = 6-8 mice per group; representative of 2 independent experiments; day 70: n = 9-10 mice per group). (D) Time C57Bl6 recipients spent swimming (mobile) in the forced swim test at days 35 and 70 after transplant (n = 8-9 mice per group). Performance of cGVHD and TCD mice in the active place avoidance task for assessment of spatial learning in 20-minute sessions across 5 days beginning at day 35 (E) or 70 (F) after transplant (n = 9-10 mice per group). Recorded parameters include improvement from day 1 to 5 expressed as a percentage (calculated based on the difference in the number of entries into the shock zone), total number of entries into the shock zone per day, and maximum time spent avoiding the shock zone per day. Data are presented as mean ± standard error of the mean. Statistics: unpaired Student t test for differences between GVHD and TCD mice (improvement) (B,D-F), and repeated measures 2-way analysis of variance followed by Bonferroni post hoc comparison (entries, avoidance time) (E-F). *P < .05, **P < .01, ****P < .0001.
cGVHD induces prolonged behavioral deficits. (A) Schematic of transplantation regime. Lethally irradiated B6D2F1 (H2b/d) recipient mice received 5 × 106 TCD BM with no T cells or with 0.5 × 106 CD3+ T cells from C57Bl/6 (H2b) donors to induce low-grade nonlethal cGVHD. (B) Schematic of transplantation regime. Ten × 106 BM with or without 5 × 106 CD3+ T cells from a BALB/c (H2d) donor were transplanted into lethally irradiated C57Bl6 (H2b) recipients. (C) Time B6D2F1 recipients spent swimming (mobile) in the forced swim test at days 35 and 70 after transplant (day 35: n = 6-8 mice per group; representative of 2 independent experiments; day 70: n = 9-10 mice per group). (D) Time C57Bl6 recipients spent swimming (mobile) in the forced swim test at days 35 and 70 after transplant (n = 8-9 mice per group). Performance of cGVHD and TCD mice in the active place avoidance task for assessment of spatial learning in 20-minute sessions across 5 days beginning at day 35 (E) or 70 (F) after transplant (n = 9-10 mice per group). Recorded parameters include improvement from day 1 to 5 expressed as a percentage (calculated based on the difference in the number of entries into the shock zone), total number of entries into the shock zone per day, and maximum time spent avoiding the shock zone per day. Data are presented as mean ± standard error of the mean. Statistics: unpaired Student t test for differences between GVHD and TCD mice (improvement) (B,D-F), and repeated measures 2-way analysis of variance followed by Bonferroni post hoc comparison (entries, avoidance time) (E-F). *P < .05, **P < .01, ****P < .0001.
cGVHD is associated with neuroinflammation, local proinflammatory cytokine production, and T-cell infiltration
Proinflammatory cytokine mediation of effector pathways is critical to the development of systemic cGVHD,33 and cytokine dysregulation is also a common feature of neuroinflammatory diseases.34 Analysis of the brain inflammatory milieu by quantitative real-time polymerase chain reaction at day 35 demonstrated Ifny, Il1b, Tnf, and Ccl2 upregulation in cGVHD mice compared with TCD controls (Figure 2A). Having observed deficits in hippocampal-dependent spatial learning, we additionally profiled the isolated hippocampus and observed similar trends (Figure 2B). At day 70, cGVHD brains demonstrated elevated Ifng and Ccl2, with Ifng also upregulated in isolated hippocampi (Figure 2A-B), findings that were recapitulated in the BALB/c into B6 model (supplemental Figure 2A-B). Notably, Tnf expression was attenuated at day 70, indicating a restricted contribution to aGVHD7 and early cGVHD (day 35) pathologies. Recipients of NZ-eGFP BM grafts supplemented with B6.RFP+ T cells were used for flow cytometric profiling of the CNS inflammatory infiltrate (Figure 2C). Digestion of GVHD brains at days 14, 35, and 70 after transplant revealed significant T-cell infiltration that comprised ∼60% of the CD45+ cells at day 14 and gradually decreased over time (Figure 2E-F). Consistent with reports in aGVHD,6 CD8+ T cells were predominant at day 14 (Figure 2G); however, over time, a gradual increase in the proportion and absolute number of CD4 T cells was noted (Figure 2G). Initial brain-infiltrating CD8+ T cells originated primarily from the donor graft T-cell compartment (Figure 2H). By day 70, however, 20% of the CD4+ T compartment was derived from BM, suggestive of prolonged low-grade infiltration (Figure 2I). To further identify parenchymal brain regions of interest and possible cellular routes of infiltration, 5 brain structures35 were investigated for the presence of RFP+ donor graft T cells (Figure 3A). Both the meninges and choroid plexus represent points of access for inflammatory infiltrate,36,37 and at day 14, significant T-cell infiltrate was observed in both of these sites (Figure 3B-C). Although T-cell numbers markedly declined in the meninges by day 35, infiltration in the choroid plexus persisted to day 70. Parenchymal localization peaked at day 35 in the cortex (Figure 3D) and hippocampus (dentate gyrus region; Figure 3E), with reductions evident in both regions by day 70. In addition, we observed a similar trend when examining the habenula nuclei, which are diversely involved in many neuromodulatory systems, regulate cognitive and motivational processes, and are known to be dysfunctional in depression.38 Collectively, these data suggest a persistent inflammatory milieu in CNS cGVHD with features distinct from those in aGVHD.
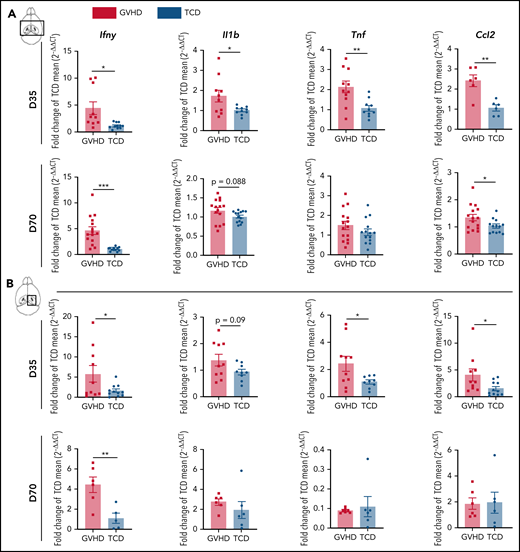
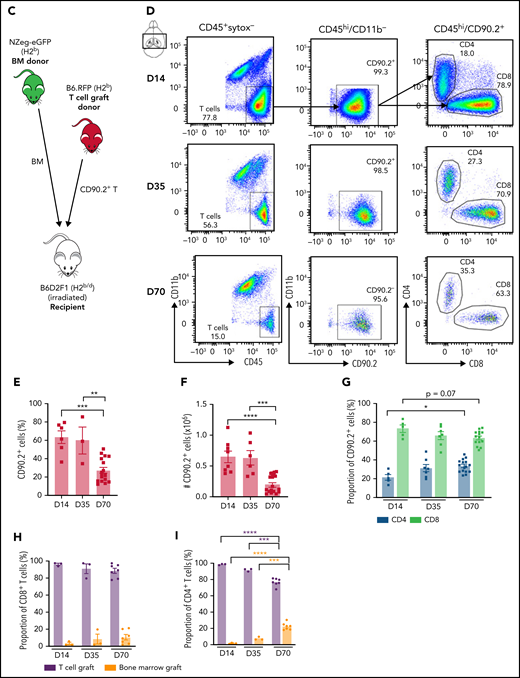
cGVHD induces T-cell infiltration and a proinflammatory cytokine profile in the brain. Messenger RNA expression of selected proinflammatory cytokines detected by quantitative real-time polymerase chain reaction in the thick coronal section (A) and hippocampus (B) of GVHD and TCD mice at days 35 and 70 after transplant (n = 6-15 mice per group; data pooled from 2 independent experiments). Expression calculated relative to the Hprt gene and reported as a fold change of the mean of the TCD group. (C) Schematic of transplantation regime. Lethally irradiated B6D2F1 recipient mice received 5 × 106 BM from C57Bl/6 donors ubiquitously expressing GFP and 0.5 × 106 sort-purified CD90.2+ T cells from C57Bl/6 donors ubiquitously expressing RFP. (D) Representative dot plots indicating gating strategy for identifying live CD90.2+ CD4 and CD8 T-cell subsets within the CD45hiCD11b− population, gated on forward and side scatter. Cells were isolated from digested coronal brain segments of transplant recipients at days 14, 35, and 70 after transplant. (E-I) Quantification of lymphocyte populations in dissociated GVHD brains at days 14, 35, and 70 after transplant. (E) CD90.2+ proportion of the CD45hiCD11b− population. (day 14: n = 6; data pooled from 2 independent experiments; day 35: n = 3; data from 1 independent experiment; day 70: n = 17; data pooled from 4 independent experiments). (F) Absolute number of CD90.2+ T cells within the CD45hiCD11b− population (day 14: n = 8; data pooled from 3 independent experiments; day 35: n = 6; data pooled from 2 independent experiments; day 70: n = 16; data pooled from 4 independent experiments). (G) Proportions of CD4+ and CD8+ T cells within the CD90.2+ population (day 14: n = 5; data pooled from 2 independent experiments; day 35: n = 7; data pooled from 2 independent experiments; day 70: n = 16; data pooled from 4 independent experiments). (H) Comparison of the proportions of CD8+ T cells derived from the T-cell (red) and BM (green) grafts (days 14 and 35: n = 3 mice per time point; day 70: n = 7; data pooled from 3 independent experiments). (I) Comparison of the proportions of CD4+ T cells derived from the T-cell (red) and BM (green) grafts (days 14 and 35: n = 3 mice per time point; day 70: n = 7; data pooled from 3 independent experiments). Data are presented as mean ± standard error of the mean. Significant differences calculated using unpaired Student t test (A-B) or 1-way analysis of variance with Tukey multiple comparisons test (E-H). *P < .05, **P < .01, ***P < .001, ****P < .0001.
cGVHD induces T-cell infiltration and a proinflammatory cytokine profile in the brain. Messenger RNA expression of selected proinflammatory cytokines detected by quantitative real-time polymerase chain reaction in the thick coronal section (A) and hippocampus (B) of GVHD and TCD mice at days 35 and 70 after transplant (n = 6-15 mice per group; data pooled from 2 independent experiments). Expression calculated relative to the Hprt gene and reported as a fold change of the mean of the TCD group. (C) Schematic of transplantation regime. Lethally irradiated B6D2F1 recipient mice received 5 × 106 BM from C57Bl/6 donors ubiquitously expressing GFP and 0.5 × 106 sort-purified CD90.2+ T cells from C57Bl/6 donors ubiquitously expressing RFP. (D) Representative dot plots indicating gating strategy for identifying live CD90.2+ CD4 and CD8 T-cell subsets within the CD45hiCD11b− population, gated on forward and side scatter. Cells were isolated from digested coronal brain segments of transplant recipients at days 14, 35, and 70 after transplant. (E-I) Quantification of lymphocyte populations in dissociated GVHD brains at days 14, 35, and 70 after transplant. (E) CD90.2+ proportion of the CD45hiCD11b− population. (day 14: n = 6; data pooled from 2 independent experiments; day 35: n = 3; data from 1 independent experiment; day 70: n = 17; data pooled from 4 independent experiments). (F) Absolute number of CD90.2+ T cells within the CD45hiCD11b− population (day 14: n = 8; data pooled from 3 independent experiments; day 35: n = 6; data pooled from 2 independent experiments; day 70: n = 16; data pooled from 4 independent experiments). (G) Proportions of CD4+ and CD8+ T cells within the CD90.2+ population (day 14: n = 5; data pooled from 2 independent experiments; day 35: n = 7; data pooled from 2 independent experiments; day 70: n = 16; data pooled from 4 independent experiments). (H) Comparison of the proportions of CD8+ T cells derived from the T-cell (red) and BM (green) grafts (days 14 and 35: n = 3 mice per time point; day 70: n = 7; data pooled from 3 independent experiments). (I) Comparison of the proportions of CD4+ T cells derived from the T-cell (red) and BM (green) grafts (days 14 and 35: n = 3 mice per time point; day 70: n = 7; data pooled from 3 independent experiments). Data are presented as mean ± standard error of the mean. Significant differences calculated using unpaired Student t test (A-B) or 1-way analysis of variance with Tukey multiple comparisons test (E-H). *P < .05, **P < .01, ***P < .001, ****P < .0001.
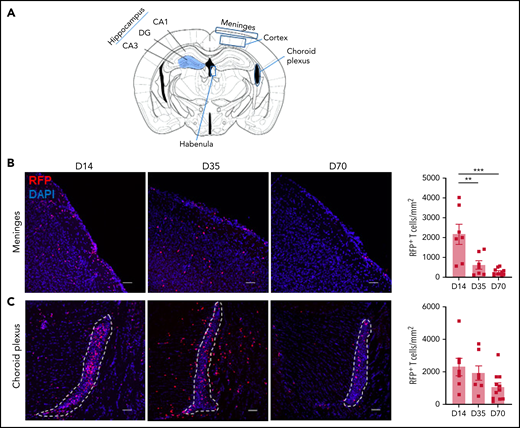
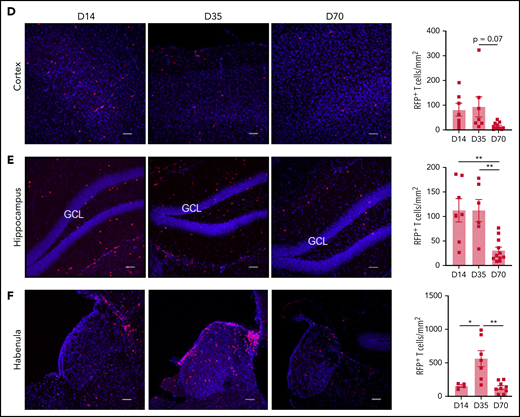
Temporal and regional changes in donor-derived T-cell infiltration. (A) Schematic of coronal brain section (based on Paxinos and Franklin35) indicating the regions examined with immunofluorescence: hippocampus, habenula, meninges, cortex, and choroid plexus (located in the lateral ventricular spaces). Blue shaded region of the hippocampus identifies the dentate gyrus (DG) containing the granule cell layer (GCL). Surrounding regions include the cornu ammonis 1 (CA1) and 3 (CA3). (B-F) Representative images of donor RFP+ T-cell infiltrate in brain regions of interest, with respective quantification at days 14, 35, and 70 after transplant: meninges (B), choroid plexus (in lateral ventricle outlined by white dashes) (C), cortex (D), hippocampus (E), and habenula (F) (day 14: n = 4-7; data pooled from 2 independent experiments; day 35: n = 6-7; data pooled from 2 independent experiments; day 70: n = 9-11; data pooled from 2 independent experiments). Original magnification ×20; scale bars, 50 μm. Data are presented as mean ± standard error of the mean. Significant differences calculated with 1-way analysis of variance with Tukey multiple comparisons test. *P < .05, **P < .01, ***P < .001.
Temporal and regional changes in donor-derived T-cell infiltration. (A) Schematic of coronal brain section (based on Paxinos and Franklin35) indicating the regions examined with immunofluorescence: hippocampus, habenula, meninges, cortex, and choroid plexus (located in the lateral ventricular spaces). Blue shaded region of the hippocampus identifies the dentate gyrus (DG) containing the granule cell layer (GCL). Surrounding regions include the cornu ammonis 1 (CA1) and 3 (CA3). (B-F) Representative images of donor RFP+ T-cell infiltrate in brain regions of interest, with respective quantification at days 14, 35, and 70 after transplant: meninges (B), choroid plexus (in lateral ventricle outlined by white dashes) (C), cortex (D), hippocampus (E), and habenula (F) (day 14: n = 4-7; data pooled from 2 independent experiments; day 35: n = 6-7; data pooled from 2 independent experiments; day 70: n = 9-11; data pooled from 2 independent experiments). Original magnification ×20; scale bars, 50 μm. Data are presented as mean ± standard error of the mean. Significant differences calculated with 1-way analysis of variance with Tukey multiple comparisons test. *P < .05, **P < .01, ***P < .001.
Microglia activation and donor-derived MHC II+ macrophage infiltration are key mediators of CNS cGVHD
Inflammatory insults to the brain induce activation of microglia as resident immune effector cells, commonly accompanied by BMDM infiltration.39,40 Having observed prolonged Ccl2 upregulation and CD4+ T-cell skewing by day 70, we sought to establish perturbations to and contributions of macrophage populations in CNS cGVHD. Using flow cytometry, we examined the temporal composition of the CD45dimCD11b+Ly6G− myeloid population in brains from of B6.Csf1r-mApple41 → Csf1r-eGFPxDBA2 F1 (MacGreen-F142) transplant recipients (Figure 4A-B). Infiltrating donor (mApple) BMDMs were notable by day 35, and by day 70, they comprised ∼50% of the CD45dimCD11b+Ly6G− cells (Figure 4C). In situ staining for Iba-1 at day 35 revealed a reactive phenotype of cortical and hippocampal host microglia (supplemental Figure 3A-D), evidenced by an amoeboid soma and thickened bushy dendrites,43 in comparison with the ramified morphology of TCD microglia. However, FACS analysis of MHC II expression demonstrated that host microglial activation was transient. In contrast, donor BMDMs retained high levels of MHC II expression and coexpressed increased levels of costimulatory markers CD80, CD86, and CD40 and the phagocytosis marker CD68 (supplemental Figure 4A-E). Confocal imaging confirmed that by day 70, in all regions examined (Figure 4F-G [hippocampus]; supplemental Figure 5 [cortex, habenula, and choroid plexus]), MHC II expression was restricted to donor BMDMs, and host microglia had returned to a homeostatic phenotype. Importantly, complementary analysis at day 100 demonstrated long-term persistence of altered microglial/macrophage populations as indicated by increases in total Iba-1+ cell numbers and donor BMDMs in the choroid plexus, hippocampus (including the CA1 and CA3 regions), and habenula of cGVHD mice in both models (Figure 5; supplemental Figures 6 and 7). Comparatively, donor BMDM infiltration through the meninges dissipated after day 35, and donor BMDM infiltration into the cortex was not significant at either time point, suggesting the early increase in Iba1+ cells likely reflects an activated-induced expansion of the host microglia.
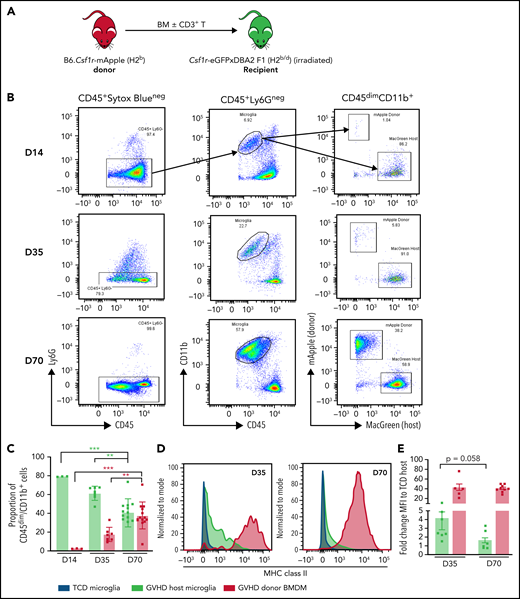
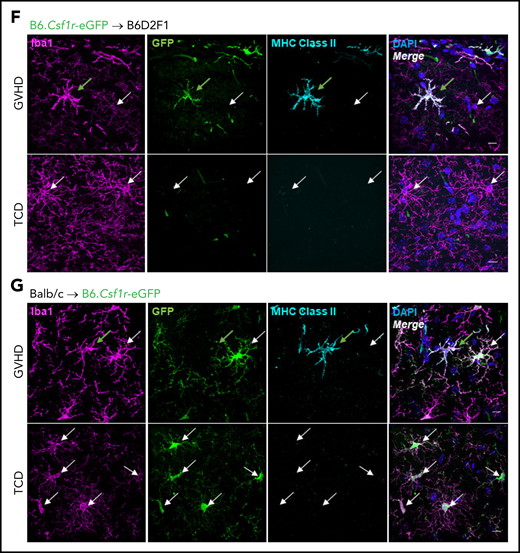
Differential expression of MHC class II by donor-derived macrophages and host microglia late after transplant. (A) Schematic of transplantation regime for the identification of donor macrophages and host microglia. Lethally irradiated DBA2xF1.Csf1r-eGFP (H2b/d) mice received 5 × 106 TCD BM with no T cells or with 0.5 × 106 CD3+ T cells from C57Bl/6.Csf1r-mApple (H2b) donors to induce low-grade nonlethal cGVHD. (B) Representative flow cytometric dot plots for the identification of donor macrophages (mApple) and host microglia (MacGreen) within the CD45dimCD11b+ population from digested coronal sections of the brains of cGVHD mice at days 14, 35, and 70 after transplant. Gated on forward and side scatter to identify live (Sytox Blue−) CD45+Ly6G− cells. (C) Proportions of host microglia and donor BMDMs within the CD45dimCD11b+ population in cGVHD brains (day 14: n = 3; day 35: n = 7; data pooled from 2 independent experiments; day 70: n = 13; data pooled from 4 independent experiments). (D) Representative histograms of MHC class II expression on TCD microglia compared with GVHD host microglia and donor BMDMs at days 35 and 70 after transplant. (E) Fold change of MHC class II mean fluorescence intensity (MFI) on GVHD host microglia and donor BMDMs relative to TCD host microglia at days 35 and 70 posttransplantation (day 35: n = 7; data pooled from 2 independent experiments; day 70: n = 9; data pooled from 2 independent experiments). (F) Representative ×100 original magnification confocal images demonstrating differential expression of MHC class II by donor macrophages (green arrows) and host microglia (white arrows) in situ in the hippocampus of GVHD and TCD mice 70 days after transplant. B6.Csf1r-eGFP donors used to facilitate identification of donor-derived macrophages (GFP+/Iba1+) compared with resident microglia (GFP−/Iba1+). Nuclei counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Original magnification ×100; scale bar, 10 μm. (G) Findings replicated in a second model of cGVHD (day 70 after transplant) where BALB/c donor BM plus CD3+ T cells were transplanted into B6.Csf1r-eGFP recipients. MHC class II expression restricted to donor Iba1+/GFP− BMDMs (green arrows) and absent from host Iba1+/GFP+ microglia (white arrows). Original magnification ×100; scale bar, 10 μm. Data presented as mean ± standard error of the mean. Statistical significance calculated by 1-way analysis of variance with multiple comparisons (C) or unpaired Student t test (E). **P < .01, ***P < .001.
Differential expression of MHC class II by donor-derived macrophages and host microglia late after transplant. (A) Schematic of transplantation regime for the identification of donor macrophages and host microglia. Lethally irradiated DBA2xF1.Csf1r-eGFP (H2b/d) mice received 5 × 106 TCD BM with no T cells or with 0.5 × 106 CD3+ T cells from C57Bl/6.Csf1r-mApple (H2b) donors to induce low-grade nonlethal cGVHD. (B) Representative flow cytometric dot plots for the identification of donor macrophages (mApple) and host microglia (MacGreen) within the CD45dimCD11b+ population from digested coronal sections of the brains of cGVHD mice at days 14, 35, and 70 after transplant. Gated on forward and side scatter to identify live (Sytox Blue−) CD45+Ly6G− cells. (C) Proportions of host microglia and donor BMDMs within the CD45dimCD11b+ population in cGVHD brains (day 14: n = 3; day 35: n = 7; data pooled from 2 independent experiments; day 70: n = 13; data pooled from 4 independent experiments). (D) Representative histograms of MHC class II expression on TCD microglia compared with GVHD host microglia and donor BMDMs at days 35 and 70 after transplant. (E) Fold change of MHC class II mean fluorescence intensity (MFI) on GVHD host microglia and donor BMDMs relative to TCD host microglia at days 35 and 70 posttransplantation (day 35: n = 7; data pooled from 2 independent experiments; day 70: n = 9; data pooled from 2 independent experiments). (F) Representative ×100 original magnification confocal images demonstrating differential expression of MHC class II by donor macrophages (green arrows) and host microglia (white arrows) in situ in the hippocampus of GVHD and TCD mice 70 days after transplant. B6.Csf1r-eGFP donors used to facilitate identification of donor-derived macrophages (GFP+/Iba1+) compared with resident microglia (GFP−/Iba1+). Nuclei counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Original magnification ×100; scale bar, 10 μm. (G) Findings replicated in a second model of cGVHD (day 70 after transplant) where BALB/c donor BM plus CD3+ T cells were transplanted into B6.Csf1r-eGFP recipients. MHC class II expression restricted to donor Iba1+/GFP− BMDMs (green arrows) and absent from host Iba1+/GFP+ microglia (white arrows). Original magnification ×100; scale bar, 10 μm. Data presented as mean ± standard error of the mean. Statistical significance calculated by 1-way analysis of variance with multiple comparisons (C) or unpaired Student t test (E). **P < .01, ***P < .001.
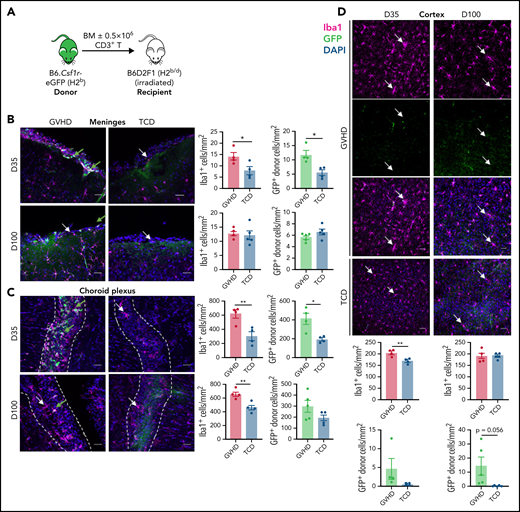
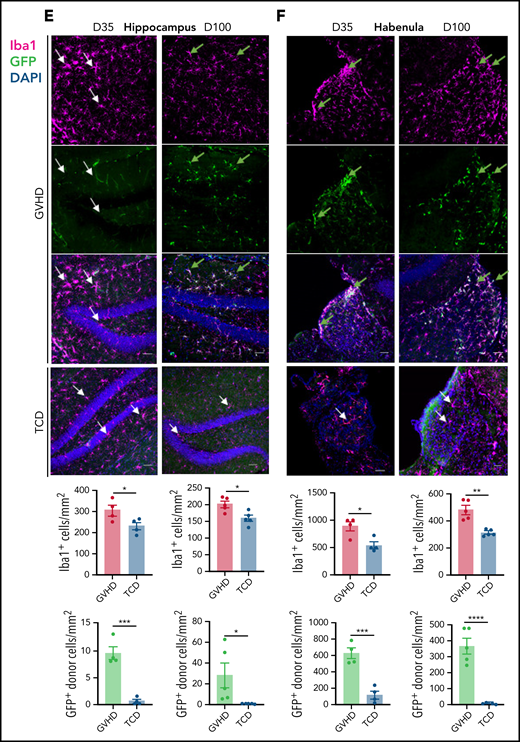
CNS cGVHD is associated with resident microglia proliferation and activation and donor-derived macrophage infiltration. (A) Allogeneic transplantation model for the delivery of 5 × 106 TCD BM from C57Bl/6 (H2b) donors expressing eGFP under the Csf1r promoter (B6.Csf1r-eGFP) with no T cells or with 0.5 × 106 CD3+ T cells into lethally irradiated B6D2F1 (H2b/d) recipients. (B-F) Representative confocal images of parenchymal brain regions of interest in GVHD and TCD mice, showing Iba1+ microglia/macrophages, GFP+ donor BMDMs, and merged images counterstained with 4’,6-diamidino-2-phenylindole (DAPI; cell nuclei). Green arrows identify donor-derived (GFP+) macrophages, and white arrows indicate resident (GFP−) microglia. Stereological quantification of each region shows the total Iba1+ cell population alongside the number of GFP+ donor BMDMs. (B) Meninges at days 35 and 100 after transplant. Original magnification ×40; scale bar, 30 μm. (C) Choroid plexus at days 35 and 100 posttransplantation. Original magnification ×40; scale bar, 30 μm. Dashed lines outline choroid plexus in the lateral ventricle adjacent to surrounding tissue. (D) Cortex at days 35 and 100 after transplant. Original magnification ×20; scale bar, 50 μm. (E) Hippocampal dentate gyrus at days 35 and 100 after transplant. Original magnification ×20; scale bar, 50 μm. (F) Habenula at days 35 and 100 after transplant. Original magnification ×40; scale bar, 30 μm. Data presented as mean ± standard error of the mean (n = 4-5 mice per group). Statistical differences calculated with unpaired Student t test. *P < .05, **P < .01, ***P < .001, ****P < .0001.
CNS cGVHD is associated with resident microglia proliferation and activation and donor-derived macrophage infiltration. (A) Allogeneic transplantation model for the delivery of 5 × 106 TCD BM from C57Bl/6 (H2b) donors expressing eGFP under the Csf1r promoter (B6.Csf1r-eGFP) with no T cells or with 0.5 × 106 CD3+ T cells into lethally irradiated B6D2F1 (H2b/d) recipients. (B-F) Representative confocal images of parenchymal brain regions of interest in GVHD and TCD mice, showing Iba1+ microglia/macrophages, GFP+ donor BMDMs, and merged images counterstained with 4’,6-diamidino-2-phenylindole (DAPI; cell nuclei). Green arrows identify donor-derived (GFP+) macrophages, and white arrows indicate resident (GFP−) microglia. Stereological quantification of each region shows the total Iba1+ cell population alongside the number of GFP+ donor BMDMs. (B) Meninges at days 35 and 100 after transplant. Original magnification ×40; scale bar, 30 μm. (C) Choroid plexus at days 35 and 100 posttransplantation. Original magnification ×40; scale bar, 30 μm. Dashed lines outline choroid plexus in the lateral ventricle adjacent to surrounding tissue. (D) Cortex at days 35 and 100 after transplant. Original magnification ×20; scale bar, 50 μm. (E) Hippocampal dentate gyrus at days 35 and 100 after transplant. Original magnification ×20; scale bar, 50 μm. (F) Habenula at days 35 and 100 after transplant. Original magnification ×40; scale bar, 30 μm. Data presented as mean ± standard error of the mean (n = 4-5 mice per group). Statistical differences calculated with unpaired Student t test. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Donor-derived macrophages and host microglia exhibit differential transcriptional profiles in the GVHD brain
Identification of a heterogeneous brain myeloid population with differential MHC II expression in cGVHD mice warranted further investigation to understand functional properties of each subset, with the intent of identifying appropriate markers or pathways for therapeutic targeting. Again, using B6.Csf1r-mApple donors and MacGreen-F1 recipients at day 70 after transplant, donor (mApple+GFP−) and host (mApple−GFP+) CD45dimCD11b+Ly6G− cells were sorted from brains of cGVHD and TCD BM control mice for bulk RNA-seq (Figure 6A). Consistent with previous reports characterizing resident microglia and infiltrating macrophages,20,44 GVHD donor BMDMs maintained a transcriptional signature distinct from GVHD host microglia, differentially expressing 5356 genes, with 2647 transcripts upregulated and 2709 downregulated (false discovery rate, <0.05; Figure 6B). A hallmark feature of microglia activation is the downregulation of signature genes.44 However, comparison of host microglia subsets from GVHD and TCD BM controls showed comparable expression of transcripts, including Cx3cr1, P2ry12, Sall1, Tmem11, and Siglech20 (Figure 6C), providing strong evidence that host microglia return to a more homeostatic state late after transplant, in line with markedly reduced MHC II expression. In contrast, these signature genes were lowly expressed or completely absent in donor BMDMs, with differential expression of CX3CR1 and P2RY12 validated by flow cytometry (Figure 6D). Additionally, donor BMDMs exhibited a distinct profile, with transcripts for Apoe, Ccr2, and Cd38, and an overlapping perivascular macrophage signature based on Cd163 and Lyve1 expression.45 Gene ontology enrichment analysis indicated a strong gene profile related to cell adhesion and migration, chemotaxis, and inflammatory responses in GVHD BMDMs compared with GVHD host microglia (Figure 6E). In line with this, GVHD donor BMDMs showed notably high expression of markers related to ECM degradation, including members of the MMP and Adam families46 (Figure 6F). The expression of a class of stimulatory CD300 molecules, involved in leukocyte response regulation,47 was almost exclusive to BMDMs (Figure 6F). This family shows similar functions to members of the triggering receptor expressed on myeloid (TREM) cell family,47 such as Trem1, also found to be upregulated in GVHD donor BMDMs and previously identified as a marker of pathogenic brain-infiltrating macrophages.44 Given our earlier findings of prolonged Ifng upregulation, we further probed this signature and found a striking upregulation of genes induced or regulated by interferon-γ (IFN-γ) signaling in the donor BMDMs (Figure 6G). Additionally, GVHD BMDMs and microglia differentially expressed genes for various cytokines, including Il1b and Il6 (Figure 6H). Coinciding with recent reports,16 resident microglia showed no in vivo Il10 expression. Differential expression of Tnf in GVHD and TCD host microglia compared with GVHD donor BMDMs is likely attributable to homeostatic glial TNF production for synaptic scaling.48 Collectively, these results delineate donor BMDMs and resident microglia as transcriptionally distinct populations, where persistent BMDM infiltration and activation mark a unique signature in the cGVHD brain.
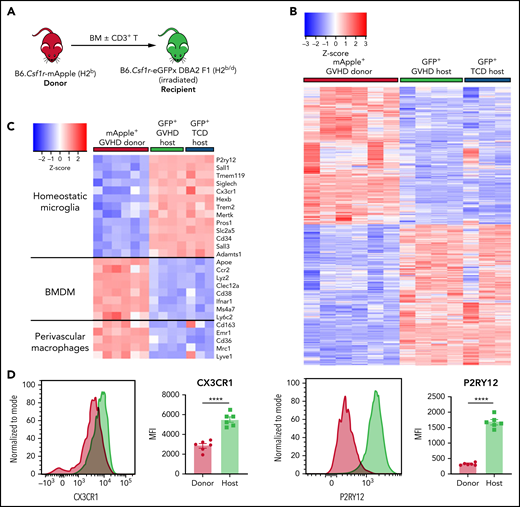
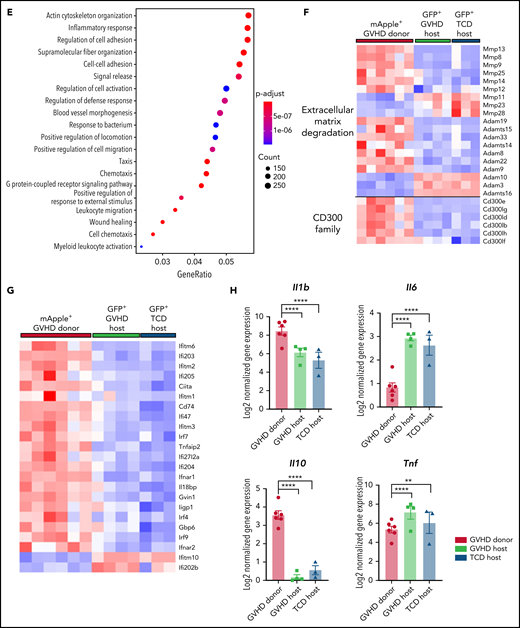
Donor-derived macrophages remain a transcriptionally and functionally distinct cell type in the CNS. (A) Schematic of transplantation regime for the identification of donor macrophages and host microglia. Lethally irradiated DBA2xF1.Csf1r-eGFP (H2b/d) mice received 5 × 106 TCD BM with no T cells or with 0.5 × 106 CD3+ T cells from C57Bl/6.Csf1r-mApple (H2b) donors to induce low-grade nonlethal cGVHD. (B) Heatmap of log2 counts-per-million (logCPM) values for differentially expressed genes (false discovery rate, <0.05) from bulk RNA-seq analysis of sort-purified infiltrating donor BMDMs compared with host microglia from GVHD brains at day 80 after transplant. Expression across each gene has been scaled so that mean expression is 0 and standard deviation is 1. Plotted alongside is the z score of the same genes from TCD host microglia (GVHD donor, n = 6; GVHD host, n = 4; TCD host, n = 3). (C) Heatmap of logCPM values from RNA-seq analysis data showing expression of genes related to characteristic cellular markers of GVHD donor BMDMs, GVHD host microglia, and TCD host microglia. Color key also applicable to panels E and G. (D) Representative histograms and enumeration of the mean fluorescence intensity of characteristic markers CX3CR1 and P2RY12 on donor BMDMs and host microglia at day 70 (n = 6 mice per group). (E) Dot plot of Gene Ontology terms from the Biological Process ontology enriched in GVHD donor BMDMs compared with GVHD host microglia. (F) Heatmap based on RNA-seq analysis data showing differentially expressed genes related to extracellular matrix (ECM) degradation and the CD300 family in GVHD donor BMDMs, GVHD host microglia, and TCD host microglia. (G) Differentially expressed genes related to IFN-γ signaling, response, and induction in GVHD donor BMDMs and host microglia from GVHD and TCD mice. (H) LogCPM values for selected cytokines from isolated cell populations in panel B. Data presented as mean ± standard error of the mean. Statistical analysis performed with unpaired Student t test (D) or using the glmLRT() function in R for all possible group comparisons (H). **P < .01, ****P < .0001.
Donor-derived macrophages remain a transcriptionally and functionally distinct cell type in the CNS. (A) Schematic of transplantation regime for the identification of donor macrophages and host microglia. Lethally irradiated DBA2xF1.Csf1r-eGFP (H2b/d) mice received 5 × 106 TCD BM with no T cells or with 0.5 × 106 CD3+ T cells from C57Bl/6.Csf1r-mApple (H2b) donors to induce low-grade nonlethal cGVHD. (B) Heatmap of log2 counts-per-million (logCPM) values for differentially expressed genes (false discovery rate, <0.05) from bulk RNA-seq analysis of sort-purified infiltrating donor BMDMs compared with host microglia from GVHD brains at day 80 after transplant. Expression across each gene has been scaled so that mean expression is 0 and standard deviation is 1. Plotted alongside is the z score of the same genes from TCD host microglia (GVHD donor, n = 6; GVHD host, n = 4; TCD host, n = 3). (C) Heatmap of logCPM values from RNA-seq analysis data showing expression of genes related to characteristic cellular markers of GVHD donor BMDMs, GVHD host microglia, and TCD host microglia. Color key also applicable to panels E and G. (D) Representative histograms and enumeration of the mean fluorescence intensity of characteristic markers CX3CR1 and P2RY12 on donor BMDMs and host microglia at day 70 (n = 6 mice per group). (E) Dot plot of Gene Ontology terms from the Biological Process ontology enriched in GVHD donor BMDMs compared with GVHD host microglia. (F) Heatmap based on RNA-seq analysis data showing differentially expressed genes related to extracellular matrix (ECM) degradation and the CD300 family in GVHD donor BMDMs, GVHD host microglia, and TCD host microglia. (G) Differentially expressed genes related to IFN-γ signaling, response, and induction in GVHD donor BMDMs and host microglia from GVHD and TCD mice. (H) LogCPM values for selected cytokines from isolated cell populations in panel B. Data presented as mean ± standard error of the mean. Statistical analysis performed with unpaired Student t test (D) or using the glmLRT() function in R for all possible group comparisons (H). **P < .01, ****P < .0001.
RNA-seq reveals molecular synaptic changes in the cGVHD hippocampus and defines a critical role for donor MHC II
To identify targetable pathways related to cellular infiltration and inflammation that may contribute to altered behavior in cGVHD mice, we conducted bulk RNA-seq on the hippocampi of GVHD and TCD mice at day 80 after transplant. After removal of outliers based on principal component analysis, downstream differential gene expression analysis identified 381 significantly differentially expressed genes (false discovery rate, <0.05; Figure 7A), with 147 genes found to be upregulated in GVHD and 207 genes downregulated. Gene set enrichment analysis found that the 10 most significantly upregulated gene sets, as identified through the Gene Ontology database, were related to general immune responses (supplemental Table 4). Confirming our previous findings of a strong IFN-γ response during cGVHD, the hallmark gene set within gene set enrichment analysis revealed significant upregulation of the IFN-induced proteins Ifi44, Ifit3, and Ifit1 in GVHD hippocampi (Figure 7B). Ingenuity pathway analysis also showed upregulation in the GVHD hippocampus of molecules related to antigen presentation (Figure 7C; supplemental Table 5), including subunits of both MHC I (Hs-T23, H2-K1, and H2-Q7) and MHC II (H2-Ab1 and H2-Dmb1), as well as the MHC class II transactivator CIITA and Ctss, required for degradation of antigenic proteins to peptides for MHC II presentation.49
As a molecular correlate for altered behavior, changes to synaptic structure and function are widely reported in various neurodegenerative and inflammatory CNS conditions.50 The Gene Ontology database identified an ontology related to synapse structure and function containing 15 genes downregulated in the GVHD hippocampus (Figure 7D). Perturbations to the expression of genes transcribing trafficking proteins (Arhgap33),51 components of the neurotransmitter release machinery (Prrt2),52 cell adhesion molecules (Nrxn2 and Nlgn2),53 and postsynaptic receptor subunits (Grin2D)54 indicate the stability, activity, and number of functional synapses may be compromised. To further investigate the premise of synapse disruption in vivo, we quantified synaptophysin (Figure 7E) and PSD-95 (data not shown) expression in the hippocampus as indicators of the pre- and postsynapse, respectively. Although no differences were evident in PSD-95 protein expression, by day 35, the GVHD hippocampus showed a trend toward reduced synaptophysin puncta density compared with TCD BM controls, which was significantly reduced at day 70. This is suggestive of a preferential disruption to the presynaptic compartment, suggesting that alterations to hippocampal synaptic structure and transmission may contribute the observed behavioral phenotype in cGVHD.
![Transcriptional changes in the hippocampus in cGVHD and attenuation of neuroinflammation by donor MHC II KO. (A) Heatmap representing log2 counts-per-million (logCPM) values of 381 differentially expressed genes (false discovery rate [FDR], <0.05) based on RNA-seq analysis of isolated hippocampi from TCD and GVHD mice 70 days after transplant (n = 3 per group). Expression across each gene has been scaled so that mean expression is 0 and standard deviation is 1. (B-C) Heatmap of logCPM values from TCD and GVHD mouse hippocampi for genes related to the IFN-γ response hallmark gene set (B) and antigen presentation (C) as identified by ingenuity pathway analysis of RNA-seq data (n = 3 per group). (D) Heatmap of logCPM values for genes involved in synaptic signaling as identified by Gene Ontology analysis (0045202). Enrichment plot from gene set enrichment analysis demonstrating downregulation of genes associated with the neuron-to-neuron synapse in GVHD compared with TCD. (E) Quantification of synaptophysin puncta expression in the hippocampus at days 35 (n = 8 mice per group; pooled from 2 independent experiments) and 70 (n = 6-11 mice per group) after transplant. (F) Schematic for the transplantation of WT (PTPxC57Bl/6) or MHC II KO.B6 (H2b) bone marrow depleted of T-cells into irradiated DBA2xF1 (H2Dd) recipients. (G) Clinical scores of transplant recipients (n = 11-12 mice per group; data pooled from 2 independent experiments). (H) Representative images of hematoxylin and eosin staining of skin from recipient mice at day 70 after transplant showing dermal thickening and loss of subcutaneous fat as evidence of scleroderma. Original magnification ×20; scale bar, 300 μm. (I) Time spent floating (immobile) and swimming (mobile) in the forced swim test (n = 4 mice per group) at day 70. (J) Representative confocal images of Iba1+ cells in the hippocampus at day 70 after transplant. Resident microglia express GFP and are indicated with white arrows. Original magnification ×20; scale bar, 50 μm. (K) Representative confocal images demonstrating morphological similarities in the resident microglia phenotype of recipients of MHC II KO TCD BM and WT TCD BM. Original magnification ×100; scale bar, 10 μm. (L-M) Messenger RNA expression of selected genes detected by quantitative real-time polymerase chain reaction in the coronal brain section (L) and hippocampus (M) of transplant recipients at day 70 after transplant. Reported as the fold change of the expression from the WT TCD BM group, calculated relative to the Hprt gene (n = 3-6 mice per group). Data are presented as mean ± standard error of the mean (SEM). Significant differences calculated using 2-way analysis of variance (G) or unpaired Student t test (E,M). *P < .05, ***P < .001. DAPI, 4’,6-diamidino-2-phenylindole; FOV, field of view; NES, normalized enrichment score.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/9/10.1182_blood.2021011671/6/m_bloodbld2021011671f7a.png?Expires=1767730151&Signature=w8SGCGRCiwGa6d4UBICABwsblR6e982SJ1WVNE4lsrQL6OZLgeGnXbiUEXku2jVaMFFoBvcTmNRV7Fq3af~q2-2lPWASi7aeLg-ygG2Omq8gdLAvcUt5zy2gkDFQSWHz3cAJ-t9PY2ZkuPlkfWOoj3aeL~j3BteyfjM0uNbQr0PR8G7hqklpdsFqoQhvFo9-HSm-sWFBbZ20ZnuKMqyFWCvvVH9VaIQVfFLpHI0G4enN-R8lCGuq1WRRW0oiIB~NOe4vYFqn4Rdhn9GB06iljnOHyBax8b2b02M~AxfV4a5AFaZA3azWTBA6UviJZGTvX2U3ZWlu21WBZ3jSHMtz6Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![Transcriptional changes in the hippocampus in cGVHD and attenuation of neuroinflammation by donor MHC II KO. (A) Heatmap representing log2 counts-per-million (logCPM) values of 381 differentially expressed genes (false discovery rate [FDR], <0.05) based on RNA-seq analysis of isolated hippocampi from TCD and GVHD mice 70 days after transplant (n = 3 per group). Expression across each gene has been scaled so that mean expression is 0 and standard deviation is 1. (B-C) Heatmap of logCPM values from TCD and GVHD mouse hippocampi for genes related to the IFN-γ response hallmark gene set (B) and antigen presentation (C) as identified by ingenuity pathway analysis of RNA-seq data (n = 3 per group). (D) Heatmap of logCPM values for genes involved in synaptic signaling as identified by Gene Ontology analysis (0045202). Enrichment plot from gene set enrichment analysis demonstrating downregulation of genes associated with the neuron-to-neuron synapse in GVHD compared with TCD. (E) Quantification of synaptophysin puncta expression in the hippocampus at days 35 (n = 8 mice per group; pooled from 2 independent experiments) and 70 (n = 6-11 mice per group) after transplant. (F) Schematic for the transplantation of WT (PTPxC57Bl/6) or MHC II KO.B6 (H2b) bone marrow depleted of T-cells into irradiated DBA2xF1 (H2Dd) recipients. (G) Clinical scores of transplant recipients (n = 11-12 mice per group; data pooled from 2 independent experiments). (H) Representative images of hematoxylin and eosin staining of skin from recipient mice at day 70 after transplant showing dermal thickening and loss of subcutaneous fat as evidence of scleroderma. Original magnification ×20; scale bar, 300 μm. (I) Time spent floating (immobile) and swimming (mobile) in the forced swim test (n = 4 mice per group) at day 70. (J) Representative confocal images of Iba1+ cells in the hippocampus at day 70 after transplant. Resident microglia express GFP and are indicated with white arrows. Original magnification ×20; scale bar, 50 μm. (K) Representative confocal images demonstrating morphological similarities in the resident microglia phenotype of recipients of MHC II KO TCD BM and WT TCD BM. Original magnification ×100; scale bar, 10 μm. (L-M) Messenger RNA expression of selected genes detected by quantitative real-time polymerase chain reaction in the coronal brain section (L) and hippocampus (M) of transplant recipients at day 70 after transplant. Reported as the fold change of the expression from the WT TCD BM group, calculated relative to the Hprt gene (n = 3-6 mice per group). Data are presented as mean ± standard error of the mean (SEM). Significant differences calculated using 2-way analysis of variance (G) or unpaired Student t test (E,M). *P < .05, ***P < .001. DAPI, 4’,6-diamidino-2-phenylindole; FOV, field of view; NES, normalized enrichment score.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/9/10.1182_blood.2021011671/6/m_bloodbld2021011671f7b.png?Expires=1767730151&Signature=Cc0w6mM6NYXtj58daOj8ErGJdNZaYl6gSYxVAZHsk-WymTHIZUdYUqe5-y9uvVfNssODxP~yahKhAeBGUFkw2OLZ1ubB52OCRd7WLA5B92KZuZJe3CjZdbanPEbpniJbF93fZXRTlEtKBn5su~OnmUzNKYmImDM61nMfvuX-mPIwD8QoAt0lrYVB~~i1LUNHVHgOOHMSaEAgQ9SLgIKUrZk2JAFpOCnBJizbUznsuR85eiIgqsS42P7VXyXXnc-DyXQlUVuY1cjYQoaUbAKZrYwP8sXlLIto4WotKt49reQ3oUGV8j95xt9SB6bn6cnIpvw0AorPMW1N7BH5aPVAEQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Transcriptional changes in the hippocampus in cGVHD and attenuation of neuroinflammation by donor MHC II KO. (A) Heatmap representing log2 counts-per-million (logCPM) values of 381 differentially expressed genes (false discovery rate [FDR], <0.05) based on RNA-seq analysis of isolated hippocampi from TCD and GVHD mice 70 days after transplant (n = 3 per group). Expression across each gene has been scaled so that mean expression is 0 and standard deviation is 1. (B-C) Heatmap of logCPM values from TCD and GVHD mouse hippocampi for genes related to the IFN-γ response hallmark gene set (B) and antigen presentation (C) as identified by ingenuity pathway analysis of RNA-seq data (n = 3 per group). (D) Heatmap of logCPM values for genes involved in synaptic signaling as identified by Gene Ontology analysis (0045202). Enrichment plot from gene set enrichment analysis demonstrating downregulation of genes associated with the neuron-to-neuron synapse in GVHD compared with TCD. (E) Quantification of synaptophysin puncta expression in the hippocampus at days 35 (n = 8 mice per group; pooled from 2 independent experiments) and 70 (n = 6-11 mice per group) after transplant. (F) Schematic for the transplantation of WT (PTPxC57Bl/6) or MHC II KO.B6 (H2b) bone marrow depleted of T-cells into irradiated DBA2xF1 (H2Dd) recipients. (G) Clinical scores of transplant recipients (n = 11-12 mice per group; data pooled from 2 independent experiments). (H) Representative images of hematoxylin and eosin staining of skin from recipient mice at day 70 after transplant showing dermal thickening and loss of subcutaneous fat as evidence of scleroderma. Original magnification ×20; scale bar, 300 μm. (I) Time spent floating (immobile) and swimming (mobile) in the forced swim test (n = 4 mice per group) at day 70. (J) Representative confocal images of Iba1+ cells in the hippocampus at day 70 after transplant. Resident microglia express GFP and are indicated with white arrows. Original magnification ×20; scale bar, 50 μm. (K) Representative confocal images demonstrating morphological similarities in the resident microglia phenotype of recipients of MHC II KO TCD BM and WT TCD BM. Original magnification ×100; scale bar, 10 μm. (L-M) Messenger RNA expression of selected genes detected by quantitative real-time polymerase chain reaction in the coronal brain section (L) and hippocampus (M) of transplant recipients at day 70 after transplant. Reported as the fold change of the expression from the WT TCD BM group, calculated relative to the Hprt gene (n = 3-6 mice per group). Data are presented as mean ± standard error of the mean (SEM). Significant differences calculated using 2-way analysis of variance (G) or unpaired Student t test (E,M). *P < .05, ***P < .001. DAPI, 4’,6-diamidino-2-phenylindole; FOV, field of view; NES, normalized enrichment score.
Transcriptional changes in the hippocampus in cGVHD and attenuation of neuroinflammation by donor MHC II KO. (A) Heatmap representing log2 counts-per-million (logCPM) values of 381 differentially expressed genes (false discovery rate [FDR], <0.05) based on RNA-seq analysis of isolated hippocampi from TCD and GVHD mice 70 days after transplant (n = 3 per group). Expression across each gene has been scaled so that mean expression is 0 and standard deviation is 1. (B-C) Heatmap of logCPM values from TCD and GVHD mouse hippocampi for genes related to the IFN-γ response hallmark gene set (B) and antigen presentation (C) as identified by ingenuity pathway analysis of RNA-seq data (n = 3 per group). (D) Heatmap of logCPM values for genes involved in synaptic signaling as identified by Gene Ontology analysis (0045202). Enrichment plot from gene set enrichment analysis demonstrating downregulation of genes associated with the neuron-to-neuron synapse in GVHD compared with TCD. (E) Quantification of synaptophysin puncta expression in the hippocampus at days 35 (n = 8 mice per group; pooled from 2 independent experiments) and 70 (n = 6-11 mice per group) after transplant. (F) Schematic for the transplantation of WT (PTPxC57Bl/6) or MHC II KO.B6 (H2b) bone marrow depleted of T-cells into irradiated DBA2xF1 (H2Dd) recipients. (G) Clinical scores of transplant recipients (n = 11-12 mice per group; data pooled from 2 independent experiments). (H) Representative images of hematoxylin and eosin staining of skin from recipient mice at day 70 after transplant showing dermal thickening and loss of subcutaneous fat as evidence of scleroderma. Original magnification ×20; scale bar, 300 μm. (I) Time spent floating (immobile) and swimming (mobile) in the forced swim test (n = 4 mice per group) at day 70. (J) Representative confocal images of Iba1+ cells in the hippocampus at day 70 after transplant. Resident microglia express GFP and are indicated with white arrows. Original magnification ×20; scale bar, 50 μm. (K) Representative confocal images demonstrating morphological similarities in the resident microglia phenotype of recipients of MHC II KO TCD BM and WT TCD BM. Original magnification ×100; scale bar, 10 μm. (L-M) Messenger RNA expression of selected genes detected by quantitative real-time polymerase chain reaction in the coronal brain section (L) and hippocampus (M) of transplant recipients at day 70 after transplant. Reported as the fold change of the expression from the WT TCD BM group, calculated relative to the Hprt gene (n = 3-6 mice per group). Data are presented as mean ± standard error of the mean (SEM). Significant differences calculated using 2-way analysis of variance (G) or unpaired Student t test (E,M). *P < .05, ***P < .001. DAPI, 4’,6-diamidino-2-phenylindole; FOV, field of view; NES, normalized enrichment score.
Given the upregulation of genes related to antigen presentation in the hippocampus of cGVHD mice, along with BMDM macrophage infiltration, we investigated whether donor MHC II expression was critical for driving the robust cGVHD neuroinflammatory profile. Allogeneic MHC II KO BM transfer (Figure 7F) induced spontaneous late cGVHD (Figure 7G) characterized by scleroderma (Figure 7H), attributable to a failure of peripheral regulatory T cells.10 Strikingly, recipients of MHC II KO TCD BM showed no increase in mobile behavior in the FST compared with recipients of wild-type (WT) TCD BM (Figure 7I). Microglia density and phenotype in all regions of the brain (Figure 7J-K; supplemental Figure 8A-D) and proinflammatory cytokine messenger RNA levels (Figure 7L-M) were similar between recipients of MHC II KO vs WT TCD BM. Furthermore, recipients of MHC II KO BM supplemented with WT T cells (supplemental Figure 8E-F) demonstrated improved behavior in the FST at day 35 (supplemental Figure 8G) compared with recipients of WT BM plus T cells. Notably, MHC II deficiency did not diminish brain Ifng messenger RNA levels (supplemental Figure 8H), indicating that IFN-γ signaling functions upstream of and may be required for55 MHC II expression by donor BMDMs to promote CNS cGVHD behavioral perturbations.
Discussion
cGVHD represents a complicated clinical entity, with neurological manifestations remaining critically understudied despite a known impact on patient quality of life.56 Difficulties in proper diagnosis and management of CNS cGVHD are perpetuated by an absence of preclinical investigation into the mechanisms underpinning cerebral pathologies and neurocognitive dysfunction observed in patients.4 Here, we describe the immune landscape in CNS cGVHD and begin to elucidate disease mediators that may represent viable therapeutic targets.
The primary clinical predictor of cGVHD is preceding aGVHD, with important distinctions in underlying pathology determining heterogeneous target organ effects.2,10 Understanding temporal CNS disease changes will be critical for informing therapeutic strategies. Recently, several studies have investigated aGVHD effects on the brain from days 7 to 21 after transplant highlighting T-cell infiltration, proinflammatory cytokine production, and microglia activation as immune mediators of cognitive deficits.6,7,30 Using our low-dose T-cell model allowed evaluation of subtle aGVHD characteristics followed by cGVHD pathology late after transplant. The main features and divergences of aGVHD and cGVHD in the CNS are summarized and compared in Table 1. We observed peak CD8+ T-cell infiltration into the brain at day 14, supporting reported findings in murine and nonhuman primate models.11,30 The progressive decline in T-cell numbers by day 70 and proportional shift to a CD4+ T-cell phenotype suggest two potential phenomena. Firstly, early immune cell infiltration may be sufficient to initiate CNS disease, but prolonged inflammation may only require low-grade infiltration. Secondly, disease mechanisms driven by donor antigen-presenting cells are prominent in the brain only in cGVHD, paralleling peripheral mechanisms and indicating a clear divergence from aGVHD pathology.57
Summarized comparison of features associated with aGVHD and cGVHD in the brain
| Feature | Time of assessment posttransplantation, d | |
|---|---|---|
| 7-216,7,11,30 | 35 and 70-100 | |
| aGVHD | cGVHD | |
| Behavioral modifications | Normal grip strength and motor coordination6 (rotarod) | Normal grip strength and motor coordination (rotarod) |
| Increased mobility in FST (d 7 and 14)30 | Increased mobility in FST (d 35 and 70) | |
| Impaired spatial learning and memory (Morris water maze task, d 21)6 | Impaired spatial learning and memory (active place avoidance task, d 35 and 70) | |
| Increased anxiety (elevated plus maze, d 14) | Intact recognition memory | |
| Impaired recognition memory (d 19)7 | Normal performance on elevated plus maze | |
| Reduced exploratory behavior (d 21)19 | Normal exploratory behavior | |
| Cytokine mediators | TNF, IL-67,30 | IFN-γ, IL-1β, CCL2 |
| T-cell infiltration dynamics | T-cell infiltration across d 7-14, high numbers of CD8+ with primarily effector memory phenotype6,7,11 | T-cell infiltration peak at d 14 with proportional skew toward CD4 phenotype by d 70 |
| Microglia/macrophages | Resident microglia activation (d 14): morphological change, increased Iba1+ cell count, increased expression of MHC II7 | Resident microglia activation (d 35): morphological change, increased total Iba1+ cell count |
| Increased frequency of CD45hiCD11b+ monocytes (donor derived) at d 147 | Increased donor-derived macrophage infiltration, highest at d 70 with loss of CD45 expression overtime; defined transcriptionally as distinct from resident microglia | |
| Gene expression | Resident microglia upregulate genes involved in antigen presentation (d 14)7 | Antigen presentation upregulated in hippocampus (d 70) |
| d 14: GVHD microglia downregulate CX3CR1 (lineage marker), indicating activation7 | d 70: GVHD resident microglia return to phenotype resembling TCD microglia, with expression of CX3CR1 and P2RY12; donor-derived macrophages maintain activation status | |
| Feature | Time of assessment posttransplantation, d | |
|---|---|---|
| 7-216,7,11,30 | 35 and 70-100 | |
| aGVHD | cGVHD | |
| Behavioral modifications | Normal grip strength and motor coordination6 (rotarod) | Normal grip strength and motor coordination (rotarod) |
| Increased mobility in FST (d 7 and 14)30 | Increased mobility in FST (d 35 and 70) | |
| Impaired spatial learning and memory (Morris water maze task, d 21)6 | Impaired spatial learning and memory (active place avoidance task, d 35 and 70) | |
| Increased anxiety (elevated plus maze, d 14) | Intact recognition memory | |
| Impaired recognition memory (d 19)7 | Normal performance on elevated plus maze | |
| Reduced exploratory behavior (d 21)19 | Normal exploratory behavior | |
| Cytokine mediators | TNF, IL-67,30 | IFN-γ, IL-1β, CCL2 |
| T-cell infiltration dynamics | T-cell infiltration across d 7-14, high numbers of CD8+ with primarily effector memory phenotype6,7,11 | T-cell infiltration peak at d 14 with proportional skew toward CD4 phenotype by d 70 |
| Microglia/macrophages | Resident microglia activation (d 14): morphological change, increased Iba1+ cell count, increased expression of MHC II7 | Resident microglia activation (d 35): morphological change, increased total Iba1+ cell count |
| Increased frequency of CD45hiCD11b+ monocytes (donor derived) at d 147 | Increased donor-derived macrophage infiltration, highest at d 70 with loss of CD45 expression overtime; defined transcriptionally as distinct from resident microglia | |
| Gene expression | Resident microglia upregulate genes involved in antigen presentation (d 14)7 | Antigen presentation upregulated in hippocampus (d 70) |
| d 14: GVHD microglia downregulate CX3CR1 (lineage marker), indicating activation7 | d 70: GVHD resident microglia return to phenotype resembling TCD microglia, with expression of CX3CR1 and P2RY12; donor-derived macrophages maintain activation status | |
IL, interleukin.
The persistent elevation of IFN-γ in the brains of cGVHD mice raises an interesting prospect given dichotomous roles for IFN-γ in transplantation and GVHD biology.21 High IFN-γ expression was maintained at day 70, despite reduced T-cell infiltration, likely indicative of an alternative source and supportive of a role for IFN-γ in controlling donor T-cell expansion in allogeneic SCT settings.58 However, in the CNS niche, IFN-γ impairs hippocampal neurogenesis and plasticity, mediated by direct action on microglia, leading to cognitive deficits.59 Spatial learning and memory, a process intimately dependent on hippocampal neurogenesis32 and homeostatic microglia function,60 were profoundly altered in cGVHD mice, particularly at day 70. This suggests that the attenuation of IFN-γ overexpression by receptor blockade, cell-specific KO, or inhibition of downstream JAK/STAT may offer benefit for reducing inflammation and consequently improving cognitive function. Selective inhibition of JAK1/2 (ruxolitinib) has improved patient responses compared with standard therapy in steroid-refractory cGVHD.55 Although exact mechanisms of action are still being studied, our data suggest the reported reductions in IFN-γ61 after ruxolitinib treatment would likely result in attenuated downstream MHC II expression and thus improve CNS cGVHD outcomes.
In parallel with clinical reports,62 our findings indicate long-term behavioral changes in cGVHD mice, in conjunction with early microglia activation, donor BMDM infiltration, and sustained molecular disruption of the synapse. Microglia are responsible for the cytokine-dependent maintenance of the CNS microenvironment and response to brain insults, but they can perpetuate inflammation during disease.63 TNF production by activated microglia drives aGVHD primarily in the cortex,7 and elevated TNF expression in our day-35 cGVHD likely reflects a continuation of this acute pathology. Although Mathew et al7 demonstrated improved behavioral outcomes with microglial TNF reduction in aGVHD mice, inherent attenuation of TNF expression and a resting microglia phenotype by day 70 in cGVHD brains with maintained behavioral deficits indicate additional and/or alternative inflammatory mediators. Striking changes to the myeloid proportions in the brain by day 70 with increased donor BMDMs, coinciding with hippocampal transcriptional signatures of IFN-γ responses and antigen presentation, with the resultant reduction in resident microglia, identify this population as a therapeutic target. BMDMs in the brain parenchyma have been reported in various CNS pathologies and are known to express a distinct transcriptional signature from that of resident microglia, although their functional contribution to disease remains controversial.64 It still remains unclear if and how perturbations to the CNS myeloid compartment contribute to the observed behavioral changes in cGVHD mice, given maintained higher cognitive functioning in both the absence of resident microglia65 and presence of infiltrating macrophages.64 However, neuroinflammation is known to contribute to persistent changes in neuronal function, leading to cognitive deficits,66 and the role for donor antigen-presenting cells in perpetuating systemic cGVHD is well defined.67 RNA-seq data revealed upregulation of genes related to ECM degradation in donor BMDMs, which are known to compromise blood-brain barrier integrity through vascular damage and disrupt neuronal signaling by altering cell-ECM interactions and modulating neurotransmitter receptor activity.44,68 cGVHD BMDM gene clusters aligning with those previously associated with neurodegenerative diseases such as Alzheimer’s69 suggest this population likely differs functionally from resident microglia, and their disruption to the brain macrophage pool may be detrimental. Dependence of microglia and macrophages on CSF-1/CSF-1R signaling18,70 offers a targetable axis for pharmaceutical depletion. Antibody blockade of CSF-1R signaling improved sclerodermatous cGVHD in preclinical mouse models,18 leading to a clinical trial to test efficacy in patients (registered at www.clinicaltrials.gov as #NCT03604692). Importantly, timing of delivery to deplete BMDM monocyte precursors and the integrity of the blood-brain barrier to determine the availability of a blocking antibody in the brain are important considerations, additionally warranting comparison with small-molecule inhibitors of CSF-1R that can access the brain parenchyma. Investigation of BMDM responses to the brain microenvironment and immune challenges will prove invaluable for modulating their function and residence in the CNS, with potential implications for disease settings beyond GVHD.
In summary, this study is the first to define features of the CNS immune landscape in cGVHD, instigated by early T-cell infiltration and late effects associated with broad infiltration of MHC II+ donor BMDMs, suggestive of pathological features distinguishable from aGVHD. Although we have investigated these features in multiple brain regions, additional studies would benefit from examining other possible mechanisms, such as a role for humoral disturbances in the hypothalamic-pituitary axis in CNS inflammation and behavior,71,72 and expanding to the peripheral nervous system to investigate neuropathic manifestations. However, given the increasing emergence of clinical reports documenting neurocognitive dysfunction in patients with cGVHD, our findings have major implications for understanding CNS pathology late posttransplantation and identifying novel targets, distinct from those in aGVHD, to pursue as therapeutic strategies.
Acknowledgments
NZeg-eGFP mice were provided by Klaus Matthaei (John Curtin School of Medical Research). The authors acknowledge the assistance of the University of Queensland School of Biomedical Sciences Imaging Facility, QIMR Animal Facility, QIMR Berghofer Flow Cytometry and Imaging Facility. The authors thank Michael Rist, Amanda Stanley, and Grace Chojnowski for cell sorting, Madeleine Flynn for graphic design, and QIMR Berghofer Scientific Services, the Genome Informatics Group, and Medical Genomics Group for their support in the RNA-seq analysis. Behavioral tests were performed at the QBI Behaviour and Surgical Facility and the QIMR Animal Facility.
This work was supported by grants 1188584 (K.P.A.M.) and 1124503 (J.V.) from the National Health and Medical Research Council; 1147499 (K.P.A.M.) from Cancer Council Queensland; and R37 AI34495, P01 AI056299, and P01 CA142109 (B.R.B.) from the National Institutes of Health and by Discovery Early Career Research Award 150101578 (J.V.) from the Australian Research Council. J.V. holds a Senior Medical Research Fellowship from the Sylvia and Charles Viertel Foundation.
Authorship
Contribution: R.C.A. designed and performed experiments, analyzed data, prepared figures, and wrote the manuscript; D.C.-C. designed and performed experiments, analyzed data and edited the manuscript; S.N.S. designed and performed experiments and analyzed data; G.T.L. performed experiments and approved the manuscript; R.L.J. analyzed data and edited the manuscript; G.Q.-R. and G.B. provided intellectual input and approved the manuscript; L.T.K. analyzed data and approved the manuscript; A.M. provided reagents and edited the manuscript; B.R.B. interpreted data and edited the manuscript; K.P.A.M. and J.V. together conceptualized the study; J.V. provided intellectual input and approved the manuscript; K.P.A.M. designed, led, and coordinated the project, performed experiments, analyzed data, and prepared and edited the manuscript.
Conflict-of-interest disclosure: B.R.B. receives remuneration as an advisor to Magenta Therapeutics and BlueRock Therapeutics and research funding from BlueRock Therapeutics and Rheos Medicines and is a cofounder of Tmunity Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Kelli P. A. MacDonald, QIMR Berghofer Medical Research Institute, 300 Herston Rd, Herston, QLD 4006, Australia; e-mail: kelli.macdonald@qimrberghofer.edu.au.
RNA-seq data available through the National Center for Biotechnology Information Gene Expression Omnibus database with accession #GSE179441.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal