

Key Points
Genetic truncation of the fibrinogen αC-region in mice results in hypofibrinogenemia from nonsense-mediated decay of mutant Fga mRNA.
Induced hypofibrinogenemia preserves hemostasis and antimicrobial function, but protects against venous thrombosis.

Abstract
Genetic variants within the fibrinogen Aα chain encoding the αC-region commonly result in hypodysfibrinogenemia in patients. However, the (patho)physiological consequences and underlying mechanisms of such mutations remain undefined. Here, we generated Fga270 mice carrying a premature termination codon within the Fga gene at residue 271. The Fga270 mutation was compatible with Mendelian inheritance for offspring of heterozygous crosses. Adult Fga270/270 mice were hypofibrinogenemic with ∼10% plasma fibrinogen levels relative to FgaWT/WT mice, linked to 90% reduction in hepatic Fga messenger RNA (mRNA) because of nonsense-mediated decay of the mutant mRNA. Fga270/270 mice had preserved hemostatic potential in vitro and in vivo in models of tail bleeding and laser-induced saphenous vein injury, whereas Fga−/− mice had continuous bleeding. Platelets from FgaWT/WT and Fga270/270 mice displayed comparable initial aggregation following adenosine 5′-diphosphate stimulation, but Fga270/270 platelets quickly disaggregated. Despite ∼10% plasma fibrinogen, the fibrinogen level in Fga270/270 platelets was ∼30% of FgaWT/WT platelets with a compensatory increase in fibronectin. Notably, Fga270/270 mice showed complete protection from thrombosis in the inferior vena cava stasis model. In a model of Staphylococcus aureus peritonitis, Fga270/270 mice supported local, fibrinogen-mediated bacterial clearance and host survival comparable to FgaWT/WT, unlike Fga−/− mice. Decreasing the normal fibrinogen levels to ∼10% with small interfering RNA in mice also provided significant protection from venous thrombosis without compromising hemostatic potential and antimicrobial function. These findings both reveal novel molecular mechanisms underpinning fibrinogen αC-region truncation mutations and highlight the concept that selective fibrinogen reduction may be efficacious for limiting thrombosis while preserving hemostatic and immune protective functions.
Introduction
Congenital hypodysfibrinogenemia is a rare autosomal disorder characterized by both altered fibrinogen structure and significantly reduced circulating fibrinogen levels. The disorder is often driven by mutations in the C-terminal region of the Aα chain (ie, residues 220-610, αC-region), including single nucleotide polymorphisms, truncations, and insertions. The clinical manifestations of αC region truncation mutations in 24 reported cases vary, but are generally associated with mild bleeding from reduced circulating fibrinogen levels.1 For example, patients with Fibrinogen Otago (lacking Aα 271-610) or Fibrinogen Marburg (lacking Aα 464-610) have significantly prolonged plasma clotting times, with circulating fibrinogen levels of 0.06 mg/mL and 0.6 mg/mL, respectively, compared with 2 to 4 mg/mL in healthy individuals.2,3 Notably, the molecular basis of the reduced fibrinogen levels is currently unclear.1
A significant complication to understanding the biology of hypodysfibrinogenemia is that 2 variables are simultaneously imposed (ie, low levels and mutant form of fibrinogen). Consequently, whether low circulating levels of fibrinogen, the mutated fibrinogen protein, or both, are tied to changes in the hemostatic and thrombotic properties of patients is an unresolved question. Analyses of patient plasma and recombinant fibrinogen have implicated the αC region in lateral aggregation of fibrin fibrils and interaction with fibrinolytic proteins such as plasminogen, tissue plasminogen activator (tPA), and antiplasmin.2-10 However, these studies are unable to recapitulate all of the complex interactions mediated by fibrinogen and other blood components (eg, platelets) that modulate hemostatic and thrombotic outcomes in the proper microenvironment (eg, within vessels under arterial or venous flow conditions).
To analyze the (patho)physiological implications and the underlying mechanisms of a genetic truncation mutation in the αC region of fibrinogen, we generated Fga270 mice that carry a stop codon at residue 271 of the Aα chain, analogous to Fibrinogen Otago.2 Similar to patients with αC region truncation mutations, Fga270/270 mice were hypodysfibrinogenemic. Studies of Fga270/270 mice were coupled with a new model of small interfering RNA (siRNA)-mediated reduction of normal fibrinogen in wild-type (WT) mice to determine whether alterations in hemostasis, thrombosis, and antimicrobial function were mechanistically coupled to low fibrinogen levels and/or the loss of the αC region.
Materials and methods
Detailed materials and methods can be found in the supplemental Methods and Materials on the Blood Web site.
Generation of Fga270 gene-targeted mice
Single-cell C57Bl/6J embryos were injected with Cas9 protein, donor single-stranded DNA replacement vector and single guide RNA targeting the mouse Fga gene within exon 5. Injected embryos were subsequently implanted into pseudo-pregnant female mice to generate mice carrying the Fga270 mutation. Homologous recombinants were identified by polymerase chain reaction (PCR) analysis followed by a diagnostic AflII restriction enzyme digest. In all animal studies, sex- and age-matched (8‐12 weeks) mice were used. Animal experiments and protocols were approved by the Animal Care and Use Committees of University of North Carolina at Chapel Hill and Michigan State University.
Fibrinogen knockdown with siRNA
Mice were administered 2 mg/kg of lipid nanoparticles containing siRNA against fibrinogen (siFga) or luciferase (siLuc). Lipid nanoparticles were prepared as previously described.11 For each experiment, the decrease in fibrinogen was confirmed by enzyme-linked immunosorbent assay (ELISA) and/or western blot. For clotting and infection studies, siRNA was administered at 10 days and 3 days before each experimental challenge to ensure stable knockdown of fibrinogen.
In vivo hemostasis and thrombosis analyses
Tail bleeding times were measured as described.12 Briefly, a 3-mm tail tip was excised from anesthetized mice and submerged in Tris-buffered saline (pH 7.5) containing 2 mM CaCl2 at 37°C until cessation of bleeding was sustained for more than 30 seconds. Laser-induced saphenous vein injury model was performed as previously described.13 Briefly, mice were injected with Alexa Fluor 488-conjugated antibodies to GPIX (2.5 μg, clone Xia.B4, Emfret Analytics) to label circulating platelets and with Alexa Fluor 647-conjugated antibodies against fibrin (2 μg/mouse; gift from Rodney Camire). Platelet and fibrin accumulation at laser injury sites were assessed by intravital microscopy. The inferior vena cava (IVC) stasis model was performed as described.14 Side branches were ligated and the lumbar branches cauterized. After 24 hours, mice were anesthetized and the thrombi were separated from the isolated IVC and weighed.
Statistical analyses
All analyses were performed using Prism 9. Comparisons of multiple groups were performed using analysis of variance (ANOVA) and Sidak multiple comparison test. Analyses of samples containing censored values were performed using the Kruskal-Wallis or Mantel-Cox tests. Analyses of survival were performed using the Kaplan-Meier method. Results were considered significant when P < .05.
Results
Fga270 mice that carry a premature termination codon in the Fga gene are hypodysfibrinogenemic
Mice carrying a truncation mutation in the αC region of fibrinogen were generated by introducing a premature termination codon in exon 5 of the Fga gene (Figure 1A). Incorporation of the mutation was confirmed by PCR analysis of genomic DNA (representative data in Figure 1B) and direct sequence analysis in 2 independent genetic lines (termed lines A and B). Plasma fibrinogen levels of Fga270/270 mice were ∼10% of FgaWT/WT mice (Figure 1C; supplemental Figure 1A-C). The truncated fibrinogen Aα chain had the expected molecular weight of ∼30 kDa by western blot analysis of plasma collected from mice of line B under reducing conditions using a fibrinogen Aα chain-specific antibody (Figure 1D). When nonreduced samples were analyzed by western blot against fibrinogen Bβ chain, the mutant fibrinogen (ie, FibAα270) had the predicted molecular weight, indicating that FibAα270 was able to assemble into a mature fibrinogen molecule (supplemental Figure 1D). Similar results were obtained with plasma from mice of line A (data not shown).
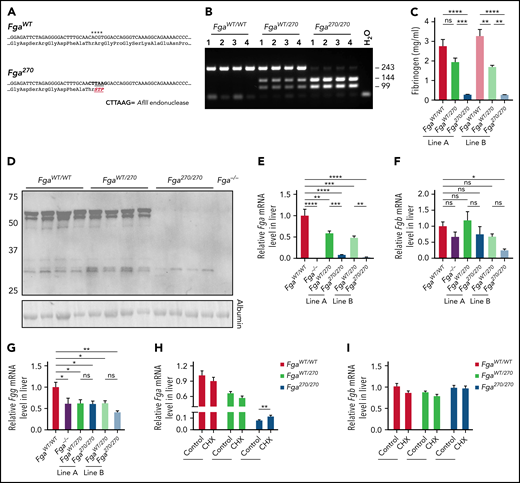
Generation of Fga270 mice that express low levels of fibrinogen with a truncated form of Aα chain after residue 270. (A) Summary of the nucleic acid substitutions introduced by Crispr-Cas9 gene editing and resulting amino acid changes for the mutated fibrinogen Aα-chain gene of the Fga270 mice. Asterisks highlight positions of the nucleotide substitutions. (B) Representative PCR analysis to establish animal genotypes of FgaWT/WT, FgaWT/270, and Fga270/270 mice. (C) ELISA measurement of plasma fibrinogen levels from FgaWT/WT, FgaWT/270, and Fga270/270 mice from 2 independent lines (n = 3-4 per genotype). One-way ANOVA was used to determine statistical significance. (D) Western blot analysis of plasma (reducing conditions) from FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice using antibodies directed against the Aα chain of fibrinogen. Analysis of hepatic mRNA levels by quantitative reverse transcriptase PCR for (E) Fga, (F) Fgb, and (G) Fgg in Fga−/− mice and 2 independent lines of Fga270 mice (n = 4 per genotype). One-way ANOVA test was used to determine statistical significance. Analysis of fibrinogen (H) Fga and (I) Fgb gene expression from primary mouse hepatocytes isolated from FgaWT/WT, FgaWT/270, and Fga270/270 mice treated with or without 100 μM cycloheximide (CHX) for 3 hours (n = 3-4 per treatment group). Two-tailed Student t test was used to determine statistical significance. *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, no statistical significance.
Generation of Fga270 mice that express low levels of fibrinogen with a truncated form of Aα chain after residue 270. (A) Summary of the nucleic acid substitutions introduced by Crispr-Cas9 gene editing and resulting amino acid changes for the mutated fibrinogen Aα-chain gene of the Fga270 mice. Asterisks highlight positions of the nucleotide substitutions. (B) Representative PCR analysis to establish animal genotypes of FgaWT/WT, FgaWT/270, and Fga270/270 mice. (C) ELISA measurement of plasma fibrinogen levels from FgaWT/WT, FgaWT/270, and Fga270/270 mice from 2 independent lines (n = 3-4 per genotype). One-way ANOVA was used to determine statistical significance. (D) Western blot analysis of plasma (reducing conditions) from FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice using antibodies directed against the Aα chain of fibrinogen. Analysis of hepatic mRNA levels by quantitative reverse transcriptase PCR for (E) Fga, (F) Fgb, and (G) Fgg in Fga−/− mice and 2 independent lines of Fga270 mice (n = 4 per genotype). One-way ANOVA test was used to determine statistical significance. Analysis of fibrinogen (H) Fga and (I) Fgb gene expression from primary mouse hepatocytes isolated from FgaWT/WT, FgaWT/270, and Fga270/270 mice treated with or without 100 μM cycloheximide (CHX) for 3 hours (n = 3-4 per treatment group). Two-tailed Student t test was used to determine statistical significance. *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, no statistical significance.
Hepatic Fga, Fgb, and Fgg messenger RNA (mRNA) levels were analyzed to determine the cause of hypofibrinogenemia in Fga270 mice. Compared with FgaWT/WT mice, Fga mRNA was decreased to 59% and 48% in FgaWT/270 mice, and 8% and 3% in Fga270/270 mice in lines A and B, respectively (Figure 1E). Hepatic Fgb mRNA levels were decreased in Fga270/270 mice only in line B, whereas Fgg mRNA levels were decreased in both FgaWT/270 and Fga270/270 mice (Figure 1F-G). To analyze for possible nonsense-mediated decay of the mutant mRNA, a protein translation-dependent process, primary hepatocytes were isolated and treated with cycloheximide.15Fga mRNA levels were significantly increased by 40% in hepatocytes of Fga270/270 mice, whereas no changes were detected in those of FgaWT/WT and FgaWT/270 mice (Figure 1H). Fgb expression levels in hepatocytes of all genotypes did not change with cycloheximide treatment (Figure 1I). As expected, no histological differences were observed in livers of Fga270 mice (supplemental Figure 2A).
Fga270 mice display postnatal success, but a fraction of Fga270/270 dams succumb to the challenge of pregnancy
Crosses of heterozygous FgaWT/270 mice produced offspring in a Mendelian distribution. For line A, 20.8% were FgaWT/WT, 54.8% were FgaWT/270, and 24.4% were Fga270/270 for the first 169 pups. Similar results were found with the first 165 pups of line B: 22.4% FgaWT/WT, 55.2% FgaWT/270, and 22.4% Fga270/270 (Table 1). The survival profile of offspring from crosses of Fga270/270 males and FgaWT/270 females produced Fga270/270 mice at 66% (P < .05) and 75% (P = .11) of expected for lines A and B, respectively. In lines A and B, 8 of 127 (6.3%) and 7 of 127 (5.1%) of Fga270/270 offspring, respectively, suffered fatal spontaneous perinatal abdominal hemorrhagic events and soft-tissue bleeds within 3 days of birth (supplemental Figure 2B). Mating of Fga270/270 dams produced mixed results. Contrary to Fga−/− dams that uniformly die of hemorrhagic events during pregnancy, 9 of 21 Fga270/270 dams survived to sustain pregnancies and deliver pups. However, Fga270/270 dams that gave birth either died of postpartum hemorrhage or were unable to nurse the newborn pups, resulting in loss of the offspring (Table 2). The pups resulting from Fga270/270 dams that died after delivery were able to survive to weaning when fostered to healthy nursing females. Adult Fga270/270 mice did not display spontaneous bleeding events nor altered survival rate compared with FgaWT/WT mice.
Analysis of postnatal survival of offspring of FgaWT/270 dams in 2 independent lines
| ♀FgaWT/270 × ♂FgaWT/270 | Line A | Line B | ||||
|---|---|---|---|---|---|---|
| Offspring genotype | FgaWT/WT | FgaWT/270 | Fga270/270 | FgaWT/WT | FgaWT/270 | Fga270/270 |
| Expected Mendelian ratio | 1 | 2 | 1 | 1 | 2 | 1 |
| Number observed (at weaning) | 35 | 92 | 41 | 37 | 91 | 37 |
| % of expected | 100% | 131% | 117% | 100% | 123% | 100% |
| ♀FgaWT/270 × ♂FgaWT/270 | Line A | Line B | ||||
|---|---|---|---|---|---|---|
| Offspring genotype | FgaWT/WT | FgaWT/270 | Fga270/270 | FgaWT/WT | FgaWT/270 | Fga270/270 |
| Expected Mendelian ratio | 1 | 2 | 1 | 1 | 2 | 1 |
| Number observed (at weaning) | 35 | 92 | 41 | 37 | 91 | 37 |
| % of expected | 100% | 131% | 117% | 100% | 123% | 100% |
| ♀FgaWT/270 × ♂Fga270/270 | Line A | Line B | ||
|---|---|---|---|---|
| Offspring genotype | FgaWT/270 | Fga270/270 | FgaWT/270 | Fga270/270 |
| Expected Mendelian ratio | 1 | 1 | 1 | 1 |
| Number observed (at weaning) | 164 | 109 | 126 | 94 |
| % of expected | 100% | 66%* | 100% | 75% |
| ♀FgaWT/270 × ♂Fga270/270 | Line A | Line B | ||
|---|---|---|---|---|
| Offspring genotype | FgaWT/270 | Fga270/270 | FgaWT/270 | Fga270/270 |
| Expected Mendelian ratio | 1 | 1 | 1 | 1 |
| Number observed (at weaning) | 164 | 109 | 126 | 94 |
| % of expected | 100% | 66%* | 100% | 75% |
P < 0.05, χ2 analysis.
Analysis of postnatal survival of offspring of Fga270/270 dams in 2 independent lines
| ♀Fga270/270 × ♂FgaWT/270 | Line A | Line B |
|---|---|---|
| Total impregnated dams | 2 | 2 |
| Gave birth and supported pups to weaning | — | — |
| Gave birth, but dam died before weaning | 1 | — |
| Gave birth, but pups died before weaning | — | — |
| Died during pregnancy | 1 | 2 |
| ♀Fga270/270 × ♂FgaWT/270 | Line A | Line B |
|---|---|---|
| Total impregnated dams | 2 | 2 |
| Gave birth and supported pups to weaning | — | — |
| Gave birth, but dam died before weaning | 1 | — |
| Gave birth, but pups died before weaning | — | — |
| Died during pregnancy | 1 | 2 |
| ♀Fga270/270 × ♂Fga270/270 | Line A | Line B |
|---|---|---|
| Total impregnated dams | 9 | 8 |
| Gave birth and supported pups to weaning | — | — |
| Gave birth, but dam died before weaning | 3 | 3 |
| Gave birth, but pups died before weaning | 2 | — |
| Died during pregnancy | 4 | 5 |
| ♀Fga270/270 × ♂Fga270/270 | Line A | Line B |
|---|---|---|
| Total impregnated dams | 9 | 8 |
| Gave birth and supported pups to weaning | — | — |
| Gave birth, but dam died before weaning | 3 | 3 |
| Gave birth, but pups died before weaning | 2 | — |
| Died during pregnancy | 4 | 5 |
Fga270/270 mice exhibit preserved hemostasis
Two distinct bleeding models were performed on Fga270 and Fga−/− mice to quantify the impact of Fga270 mutation on hemostasis. In a tail bleeding assay, FgaWT/WT and FgaWT/270 mice rapidly achieved hemostasis and stopped bleeding in 100.7 ± 14.2 and 91.5 ± 10.1 seconds, respectively. Brief rebleeding was observed in each of FgaWT/WT and FgaWT/270 mice. Notably, 5/6 Fga270/270 mice achieved hemostasis with one mouse bleeding during the entire 5-minute observation period (Figure 2A). In contrast, none of the Fga−/− mice achieved hemostasis, consistent with previous reports.16 To determine whether 10% of circulating normal fibrinogen was sufficient to maintain hemostasis, tail bleeding assays were performed on mice treated with siFga or siLuc. With this model, circulating fibrinogen was successfully reduced to ∼10% in siFga-treated mice (supplemental Figure 3). Notably, both siLuc- and siFga-treated mice readily achieved hemostasis. Using a laser-induced saphenous vein bleeding model in which each injury site was challenged 3 times, bleeding times and platelet/fibrin accumulation were quantified supplemental Video 1. Bleeding times were comparable among mice of all Fga270 genotypes, but were significantly increased for Fga−/− mice (Figure 2B). Three-dimensional reconstructions of hemostatic plugs highlighted increased extravascular platelet accumulation in Fga−/− mice (Figure 2C; supplemental Video 2). Total platelet accumulation was increased for both Fga270/270 and Fga−/− mice (Figure 2C-D). Fibrin accumulation at the injury sites was reduced in FgaWT/270 mice compared with FgaWT/WT mice (Figure 2C,E). Interestingly, fibrin accumulation in Fga270/270 mice was initially low following the first injury, but progressively increased after the second and third injuries.
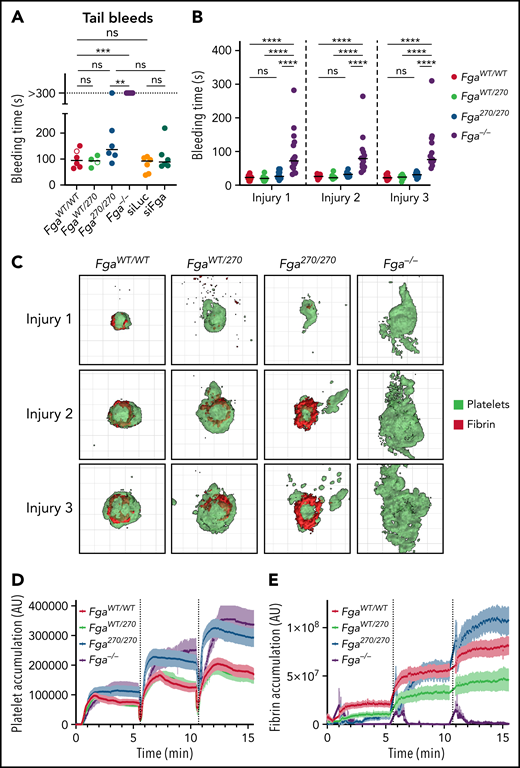
Fga270/270 and siFga-treated mice achieve hemostasis following vascular injury. (A) Time to cessation of bleeding (sustained >30 seconds) of FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice as well as siLuc- and siFga-treated mice following 3-mm excision of the distal portion of the tail. Horizontal bars indicate mean times for each group. Note that 1 of 6 Fga270/270 mice and 6 of 6 Fga−/− mice did not stop bleeding during the 5-minute evaluation period. Open dots represent mice in which rebleeding episodes occurred and the values indicate total bleeding time. Kaplan-Meier analysis was used to determine statistical significance. (B) Time to cessation of bleeding of FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice following 3 consecutive laser-induced saphenous injuries (n = 14-16 per genotype). Two-way ANOVA was used to determine statistical significance. (C) Representative 3-dimensional reconstruction of injury sites depicting the intravascular view 5 minutes following each injury. Each grid box = 50 μm × 50 μm. Quantification of (D) platelet and (E) fibrin accumulation at the site of injury over the course of 3 injuries. **P < .01, ***P < .001, ****P < .0001. ns, no statistical significance.
Fga270/270 and siFga-treated mice achieve hemostasis following vascular injury. (A) Time to cessation of bleeding (sustained >30 seconds) of FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice as well as siLuc- and siFga-treated mice following 3-mm excision of the distal portion of the tail. Horizontal bars indicate mean times for each group. Note that 1 of 6 Fga270/270 mice and 6 of 6 Fga−/− mice did not stop bleeding during the 5-minute evaluation period. Open dots represent mice in which rebleeding episodes occurred and the values indicate total bleeding time. Kaplan-Meier analysis was used to determine statistical significance. (B) Time to cessation of bleeding of FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice following 3 consecutive laser-induced saphenous injuries (n = 14-16 per genotype). Two-way ANOVA was used to determine statistical significance. (C) Representative 3-dimensional reconstruction of injury sites depicting the intravascular view 5 minutes following each injury. Each grid box = 50 μm × 50 μm. Quantification of (D) platelet and (E) fibrin accumulation at the site of injury over the course of 3 injuries. **P < .01, ***P < .001, ****P < .0001. ns, no statistical significance.
FibAα270 supports unstable platelet aggregation in vitro
To determine whether the Fga270 mutation alters platelet biology, aggregation studies were performed in vitro using platelet-rich plasma. Although aggregation of Fga−/− platelets was markedly impaired when activated with adenosine 5′-diphosphate (ADP), FgaWT/WT, FgaWT/270, and Fga270/270 platelets aggregated at similar rates. However, Fga270/270 platelets disaggregated faster compared with FgaWT/WT and FgaWT/270 platelets (Figure 3A-B). Platelets from siFga-treated mice displayed a similar profile to that of Fga270/270 platelets with an initial aggregation rate similar to platelets of siLuc-treated mice, but faster disaggregation following ADP stimulation (Figure 3C). Aggregation patterns were virtually identical for FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− platelets when stimulated with protease-activated receptor 4 activating peptide (Figure 3D). Platelet adhesion to collagen-coated surfaces under venous flow conditions was measured in a microfluidics chamber system by flowing heparinized whole blood obtained from FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice. FgaWT/WT platelets rapidly formed large and relatively stable thrombi. This phenotype was similar but less pronounced in FgaWT/270 and Fga270/270 blood, whereas Fga−/− blood formed initial thrombi that were completely washed away (Figure 3E-F; supplemental Video 3). Changes in platelet adhesion and aggregation were not from differences in platelet adhesion and spreading to purified FibAαWT and FibAα270 (Figure 3G; supplemental Figure 4) nor membrane expression of GPIIb/IIIa, GPIX, and GPIbα (Figure 3H-J).
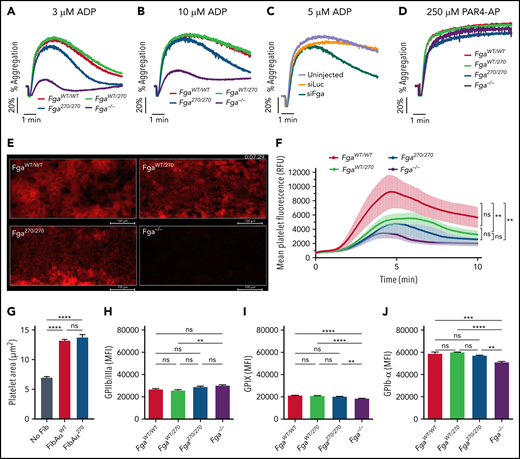
Fga270/270 platelets form weaker aggregates in response to ADP stimulation. Representative aggregation traces of platelet-rich plasma collected from FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice following stimulation with (A) 3 μM ADP or (B) 10 μM ADP (n = 4 per genotype). (C) Representative aggregation traces of platelet-rich plasma collected from untreated or siLuc- or siFga- treated mice following stimulation with 5 μM ADP (n = 4-5 per group). (D) Representative aggregation traces of platelet-rich plasma collected from FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice following stimulation with 250 μM of protease-activated receptor 4 activating peptide (PAR4-AP) (n = 4 per genotype). (E) Representative images and (F) quantification of platelet adhesion (indicated in red) to collagen coated surface at venous (400 seconds−1) shear rate using heparinized whole blood from FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice (n = 4 per genotype). (G) Adhesion and spreading of Fga−/− platelets on uncoated or FibAαWT- or FibAα270- coated coverslips following stimulation with 50 μM of ADP for 30 minutes (n = 3 per fibrinogen). One-way ANOVA was used to determine statistical significance. Expression of (H) GPIIb/IIIa, (I) GPIX, and (J) GPIbα on platelet membrane of FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice (n = 5 per genotype). One-way ANOVA was used to determine statistical significance. **P < .01, ****P < .0001. ns, no statistical significance.
Fga270/270 platelets form weaker aggregates in response to ADP stimulation. Representative aggregation traces of platelet-rich plasma collected from FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice following stimulation with (A) 3 μM ADP or (B) 10 μM ADP (n = 4 per genotype). (C) Representative aggregation traces of platelet-rich plasma collected from untreated or siLuc- or siFga- treated mice following stimulation with 5 μM ADP (n = 4-5 per group). (D) Representative aggregation traces of platelet-rich plasma collected from FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice following stimulation with 250 μM of protease-activated receptor 4 activating peptide (PAR4-AP) (n = 4 per genotype). (E) Representative images and (F) quantification of platelet adhesion (indicated in red) to collagen coated surface at venous (400 seconds−1) shear rate using heparinized whole blood from FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice (n = 4 per genotype). (G) Adhesion and spreading of Fga−/− platelets on uncoated or FibAαWT- or FibAα270- coated coverslips following stimulation with 50 μM of ADP for 30 minutes (n = 3 per fibrinogen). One-way ANOVA was used to determine statistical significance. Expression of (H) GPIIb/IIIa, (I) GPIX, and (J) GPIbα on platelet membrane of FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice (n = 5 per genotype). One-way ANOVA was used to determine statistical significance. **P < .01, ****P < .0001. ns, no statistical significance.
Comparative analyses of platelet lysates and plasma revealed that the fibrinogen content in Fga270/270 platelets was ∼30% of that in FgaWT/WT platelets (Figure 4A-B). Fibrinogen levels in Fga270/270 plasma from the same set of mice were ∼10% compared with FgaWT/WT plasma (Figure 4E-F), consistent with ELISA measurements (Figure 1C). The fibronectin content in Fga270/270 platelets was increased approximately ninefold compared with FgaWT/WT platelets (Figure 4C), consistent with previous studies indicating elevated fibronectin levels in Fga−/− platelets.17 There were no significant genotype-dependent differences in plasma fibronectin levels or platelet or plasma von Willebrand factor (VWF) levels (Figure 4D,G-H). The changes in protein content in Fga270/270 platelets appear to be driven primarily by hypofibrinogenemia. In siFga-treated mice, reduction of plasma fibrinogen to ∼4% resulted in decrease of intraplatelet fibrinogen content to ∼16% relative to siLuc-treated mice (Figure 4I-J,M-N). There was a trend toward an increase in fibronectin levels in platelets of siFga-treated mice, but not in plasma fibronectin levels, and platelet or plasma VWF levels (Figure 4K-L,O-P).
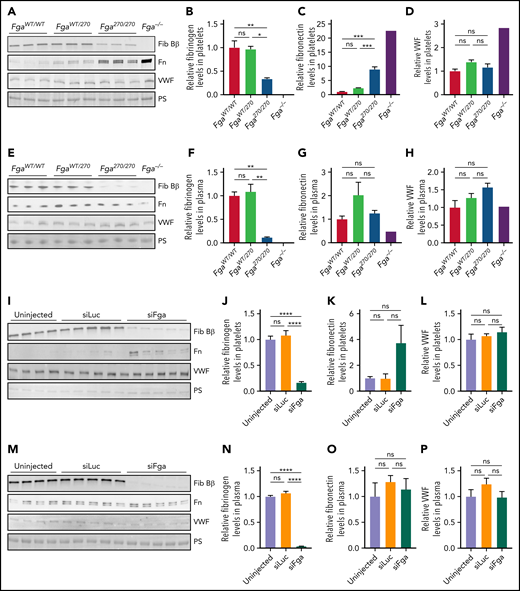
Fga270/270 platelets contain higher levels of fibrinogen relative to fibrinogen levels in plasma. (A-D) Platelet lysates and (E-H) plasma harvested from FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice using antibodies against fibrinogen Bβ-chain (Fib Bβ), fibronectin (Fn), and VWF. Ponceau S staining of albumin (PS) was used as a loading control (n = 3 per genotype). (I-L) Platelet lysates and (M-P) plasma harvested from untreated or siLuc- or siFga-treated mice using antibodies against fibrinogen Bβ-chain, fibronectin, and VWF. Ponceau S staining of albumin was used as a loading control (n = 4-5 per treatment). One-way ANOVA test was used to determine statistical significance. *P < .05, **P < .01, ****P < .0001. ns, no statistical significance.
Fga270/270 platelets contain higher levels of fibrinogen relative to fibrinogen levels in plasma. (A-D) Platelet lysates and (E-H) plasma harvested from FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice using antibodies against fibrinogen Bβ-chain (Fib Bβ), fibronectin (Fn), and VWF. Ponceau S staining of albumin (PS) was used as a loading control (n = 3 per genotype). (I-L) Platelet lysates and (M-P) plasma harvested from untreated or siLuc- or siFga-treated mice using antibodies against fibrinogen Bβ-chain, fibronectin, and VWF. Ponceau S staining of albumin was used as a loading control (n = 4-5 per treatment). One-way ANOVA test was used to determine statistical significance. *P < .05, **P < .01, ****P < .0001. ns, no statistical significance.
Fga270/270 mice display delayed clotting and impaired fibrinolysis in vitro
The impact of the Fga270 mutation on clotting parameters was determined. First, thrombin time (TT), prothrombin time (PT) and activated partial thromboplastin time (APTT) were measured with platelet-poor plasma. FgaWT/WT and FgaWT/270 plasma had comparable TT, PT, and APTT clotting times (Figure 5A-C). Fga270/270 plasma had prolonged clotting times in each assay, and some individual samples failed to clot. When FgaWT/WT plasma was diluted to 10% with Fga−/− plasma (10% FgaWT/WT) to match the Fga270/270 plasma fibrinogen concentration, there was a modest, but statistically significant delay in clotting time for TT and PT (Figure 5A-B). Significant differences between 10% FgaWT/WT and Fga270/270 were observed for TT and PT, suggesting that both the low fibrinogen levels and the mutant protein were contributing to prolonged clotting times in Fga270/270 samples. Both Fga270/270 and 10% FgaWT/WT plasma failed to clot in most APTT samples (Figure 5C). Fga−/− plasma failed to form clots over the entire observation periods for each assay. The turbidity profiles were significantly lower in Fga270/270 and 10% FgaWT/WT plasma compared with FgaWT/WT and FgaWT/270 plasma (Figure 5D). Thrombin generation of FgaWT/WT, FgaWT/270, and Fga270/270 plasma produced identical curves with no differences in lag time, time to peak, peak thrombin concentration, or endogenous thrombin potential (Figure 5E-I). Plasmin generation18 was undetectable in Fga270/270 plasma. Whereas lag time and time to peak of 10% FgaWT/WT plasma were similar to FgaWT/WT plasma, peak plasmin concentration and endogenous plasmin potential were significantly diminished (Figure 5J-N), suggesting a contribution of both the low fibrinogen levels and the Fga270 mutation.
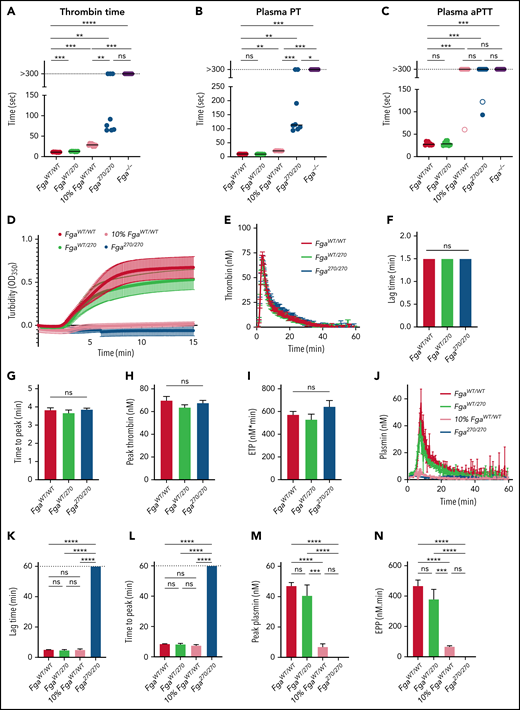
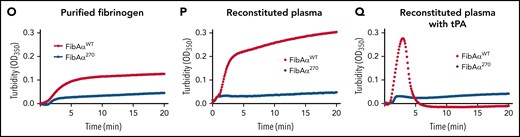
Altered clotting with Fga270/270 plasma and FibAα270 protein. (A) TT, (B) PT, and (C) APTT of FgaWT/WT, FgaWT/270, 10% FgaWT/WT, Fga270/270, and Fga−/− mice (n = 4-6 per genotype). Kaplan-Meier analysis was used to determine statistical significance. (D) Turbidity analysis of FgaWT/WT, FgaWT/270, 10% FgaWT/WT, and Fga270/270 plasma (n = 3-6 per genotype). (E) Representative thrombin generation curves and (F-I) associated parameters, and (J) representative plasmin generation curves and (K-N) associated parameters (n = 3-4 per genotype). One-way ANOVA was used to determine statistical significance. Turbidity analysis using (O) 2 mg/mL of purified FibAαWT and FibAα270 fibrinogen in buffered system and 1 mg/mL of FibAαWT and FibAα270 fibrinogen reconstituted in Fga−/− plasma in the (P) absence or (Q) presence of tPA (n = 1 per group). *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, no statistical significance.
Altered clotting with Fga270/270 plasma and FibAα270 protein. (A) TT, (B) PT, and (C) APTT of FgaWT/WT, FgaWT/270, 10% FgaWT/WT, Fga270/270, and Fga−/− mice (n = 4-6 per genotype). Kaplan-Meier analysis was used to determine statistical significance. (D) Turbidity analysis of FgaWT/WT, FgaWT/270, 10% FgaWT/WT, and Fga270/270 plasma (n = 3-6 per genotype). (E) Representative thrombin generation curves and (F-I) associated parameters, and (J) representative plasmin generation curves and (K-N) associated parameters (n = 3-4 per genotype). One-way ANOVA was used to determine statistical significance. Turbidity analysis using (O) 2 mg/mL of purified FibAαWT and FibAα270 fibrinogen in buffered system and 1 mg/mL of FibAαWT and FibAα270 fibrinogen reconstituted in Fga−/− plasma in the (P) absence or (Q) presence of tPA (n = 1 per group). *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, no statistical significance.
In turbidity assays, FibAα270 produced substantially lower turbidity profiles using purified protein in a buffer system (Figure 5O) or in reconstituted plasma (Figure 5P), suggesting an altered FibAα270 clot structure relative to FibAαWT clots. FibAαWT reconstituted in Fga−/− plasma formed clots that were readily lysed within 5 minutes by tPA, whereas no lysis were observed with FibAα270 clots (Figure 5Q). In a TT assay, FibAαWT formed stable fibrin clots, whereas FibAα270 was unable to stop the movement of the magnetic beads within the 180-second observation period, even though gel-like fibrin was present upon examination after the assay (not shown). An altered clot structure was confirmed by scanning electron microscope analyses revealing FibAα270 clots were generally less dense with substantial heterogeneity in fiber thickness (supplemental Figure 5).
Fga270/270 mice are protected from venous thrombosis
To determine the potential impact of the Fga270 mutation on thrombus formation, mice were subjected to an IVC stasis model of venous thrombosis. All FgaWT/WT and FgaWT/270 mice developed thrombi 24 hours after IVC ligation (Figure 6A-B). In contrast, neither Fga270/270 nor Fga−/− mice developed thrombi. The platelet count of Fga270/270 and Fga−/− mice at baseline were slightly lower than FgaWT/WT mice (supplemental Table 1). To determine if hypofibrinogenemia alone was protective against venous thrombosis, mice were treated with siLuc or siFga to deplete circulating fibrinogen before IVC stasis (Figure 6C). Although all 5 siLuc-treated mice developed thrombi, only 2 of 6 siFga-mice developed thrombi that were smaller than those of siLuc-treated mice (Figure 6D). Factor XIIIa (FXIIIa)-mediated crosslinking of fibrin Aα chains contributes significantly to venous thrombosis.19,20 The rate of FXIIIa activation was similar between FgaWT/WT and Fga270/270 plasma (Figure 6E). Importantly, although α chain in FgaWT/WT plasma was readily crosslinked to high-molecular-weight polymers, α-chain high-molecular-weight polymers were not detected in Fga270/270 plasma (Figure 6F). Collectively, these findings are consistent with the concept that the Fga270 mutation is protective against venous thrombosis because of both hypofibrinogenemia and the loss of αC-region.
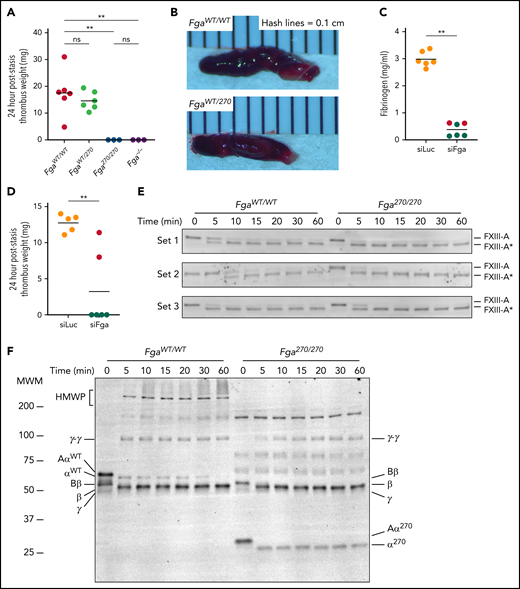
Fga270/270 and siFga-treated mice are protected from venous thrombosis. (A) Thrombus weights from FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice 24 hours after IVC ligation (stasis model). Kruskal-Wallis test was used to determine statistical significance. (B) Representative images of thrombi isolated 24 hours after IVC ligation. (C) Circulating fibrinogen levels of siLuc- and siFga-treated mice immediately before the IVC ligation, measured by ELISA. Red dots indicate mice that later developed occlusive thrombi. (D) Thrombus weights from siLuc- and siFga-treated mice 24 hours after ligation. Mantel-Cox test was used to determine statistical significance. Time course of (E) FXIII activation and (F) fibrin crosslinking in FgaWT/WT and Fga270/270 plasma. HMWP, high-molecular-weight polymer; MWM, molecular weight marker. **P < .01. ns, no statistical significance.
Fga270/270 and siFga-treated mice are protected from venous thrombosis. (A) Thrombus weights from FgaWT/WT, FgaWT/270, Fga270/270, and Fga−/− mice 24 hours after IVC ligation (stasis model). Kruskal-Wallis test was used to determine statistical significance. (B) Representative images of thrombi isolated 24 hours after IVC ligation. (C) Circulating fibrinogen levels of siLuc- and siFga-treated mice immediately before the IVC ligation, measured by ELISA. Red dots indicate mice that later developed occlusive thrombi. (D) Thrombus weights from siLuc- and siFga-treated mice 24 hours after ligation. Mantel-Cox test was used to determine statistical significance. Time course of (E) FXIII activation and (F) fibrin crosslinking in FgaWT/WT and Fga270/270 plasma. HMWP, high-molecular-weight polymer; MWM, molecular weight marker. **P < .01. ns, no statistical significance.
Fga270/270 mice have preserved fibrinogen-dependent antimicrobial function
To determine whether the Fga270 mutation influences fibrin(ogen)-dependent activities independent of hemostasis and thrombosis, we analyzed fibrin(ogen)-mediated antimicrobial function. FgaWT/WT, Fga270/270, and Fga−/− mice were challenged with an intraperitoneal infection of Staphylococcus aureus USA300, a methicillin-resistant S aureus strain. All Fga−/− mice succumbed to the infection within 78 hours, whereas 50% of FgaWT/WT mice and 62% of Fga270/270 mice survived the 1-week observation period (Figure 7A). In a parallel study evaluating fibrinogen-dependent bacterial clearance, FgaWT/WT mice eliminated ∼99% of the initial inoculum from the peritoneal cavity within 1 hour of infection, whereas Fga−/− failed to clear the microbe (Figure 7B-C).12,21 Notably, Fga270/270 mice cleared the microbes from the peritoneal cavity as efficiently as FgaWT/WT mice. The loss of fibrinogen also promoted bacterial dissemination with significantly higher colony-forming units (CFUs) in blood and lung, and a trend of higher CFU in the heart of infected Fga−/− mice relative to FgaWT/WT mice (Figure 7D-F). Importantly, the CFU burden in the blood, lung, and heart tissue of Fga270/270 mice was comparable to FgaWT/WT mice. Similarly, reduction of circulating normal fibrinogen to ∼10% (supplemental Figure 6) was sufficient to retain fibrin(ogen)-mediated antimicrobial function because siLuc- and siFga-treated mice challenged with S aureus had no differences in bacterial CFUs in the peritoneal lavage, blood, lungs, and heart (Figure 7G-J).
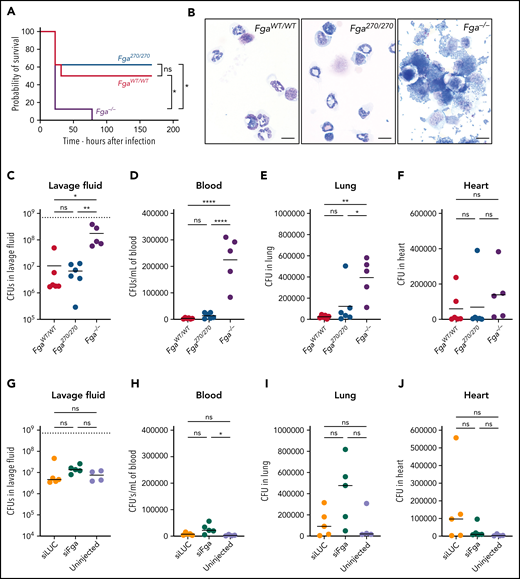
Fga270/270 and siFga-treated mice have preserved fibrinogen-dependent antimicrobial function. (A) Kaplan-Meier analysis of survival of FgaWT/WT, Fga270/270, and Fga−/− mice after intraperitoneal infection with 0.7 × 109 CFUs of S aureus (n = 8 per genotype). (B) Representative photomicrographs of cytospin preparations of peritoneal lavage fluid 1 hour after intraperitoneal infection with 0.7 × 109 CFUs of S aureus from FgaWT/WT, Fga270/270, and Fga−/− mice (n = 6 per genotype). Note the cell-associated and free bacteria in samples from Fga−/− mice that are absent in samples from FgaWT/WT and Fga270/270 mice. Scale bar: 10 μm. CFUs within (C) lavage fluid, (D) blood, (E) lung homogenates, and (F) heart homogenates after intraperitoneal infection of S aureus in FgaWT/WT, Fga270/270, and Fga−/− mice. CFUs within (G) lavage fluid, (H) blood, (I) lung homogenates, and (J) heart homogenates after intraperitoneal infection with S aureus in untreated, siLuc- and siFga-treated mice. The dashed lines in panels C and G indicate the initial inoculum dose in CFUs. One-way ANOVA test was used to determine statistical significance. *P < .05, **P < .01, ****P < .0001. ns, no statistical significance.
Fga270/270 and siFga-treated mice have preserved fibrinogen-dependent antimicrobial function. (A) Kaplan-Meier analysis of survival of FgaWT/WT, Fga270/270, and Fga−/− mice after intraperitoneal infection with 0.7 × 109 CFUs of S aureus (n = 8 per genotype). (B) Representative photomicrographs of cytospin preparations of peritoneal lavage fluid 1 hour after intraperitoneal infection with 0.7 × 109 CFUs of S aureus from FgaWT/WT, Fga270/270, and Fga−/− mice (n = 6 per genotype). Note the cell-associated and free bacteria in samples from Fga−/− mice that are absent in samples from FgaWT/WT and Fga270/270 mice. Scale bar: 10 μm. CFUs within (C) lavage fluid, (D) blood, (E) lung homogenates, and (F) heart homogenates after intraperitoneal infection of S aureus in FgaWT/WT, Fga270/270, and Fga−/− mice. CFUs within (G) lavage fluid, (H) blood, (I) lung homogenates, and (J) heart homogenates after intraperitoneal infection with S aureus in untreated, siLuc- and siFga-treated mice. The dashed lines in panels C and G indicate the initial inoculum dose in CFUs. One-way ANOVA test was used to determine statistical significance. *P < .05, **P < .01, ****P < .0001. ns, no statistical significance.
Discussion
In this study, we generated Fga270/270 mice with a truncated αC region of fibrinogen and confirmed these mice have severe hypodysfibrinogenemia, consistent with reports of patients with homozygous truncation mutations within the αC region of fibrinogen, particularly within Aα 220-447.1,22-24 Low circulating fibrinogen levels in patients were previously attributed to decreased assembly, secretion and/or plasma half-life of the mutant fibrinogen protein, based on observations of low ratios of mutant:WT Aα chains in plasma of heterozygous pateints.1-372526 An important caveat of previous reports is that analyses of patient hepatic fibrinogen mRNA levels are not available because liver tissue biopsies are not feasible. Our findings indicate that reduced fibrinogen in circulation of Fga270/270 mice was due to elimination of mutant mRNA. Indeed, Fga270/270 mice, which encode a mutation comparable to Fibrinogen Otago, have Fga mRNA levels at ∼10% of normal, matching the reduction in circulating protein. The observed rescue of Fga270/270 mRNA with cycloheximide was consistent with studies of truncation mutations in other coagulation factors, including Fibrinogen Shizuoka III and Kanazawa II, FXIII, and tissue factor.27-30 Notably, whereas previous analyses used transgene expression in heterologous cell lines, we analyzed nonsense-mediated decay of an endogenous coagulation factor gene using primary cells.
Despite profound hypofibrinogenemia, Fga270/270 mice had preserved hemostatic potential that could be appreciated from birth. The expected Mendelian distribution of Fga270/270 mice were observed at weaning from crosses of heterozygous parents. This is notable because a substantial number of Fga−/− mice do not survive to weaning but instead suffer fatal abdominal hemorrhages within 3 days of birth.16 Intriguingly, we observed a modest reduction in the number of Fga270/270 mice at weaning in crosses of Fga270/270 sires with FgaWT/270 dams. The reason for this modest difference based on breeding strategy is unknown. The pregnancy profile in Fga270/270 dams is also markedly distinct from that of Fga−/− animals. Although all Fga−/− dams die of hemorrhages during pregnancy, a subset of Fga270/270 dams were able to sustain pregnancies and deliver pups, indicating sufficient fibrinogen concentration and function required for pregnancy.16 The occasional miscarriages and postpartum hemorrhages of Fga270/270 dams mimic observations of miscarriages in Fibrinogen Otago and Fibrinogen Mannheim V patients, and postpartum hemorrhages in Fibrinogen Perth and Fibrinogen Lincoln patients.2,6,7
Functional studies in Fga270/270 mice and the siFga model supported a conclusion of a mildly perturbed, but largely preserved hemostatic potential. Fga270/270 and siFga-treated mice, but not Fga−/− mice, achieved hemostasis in the tail bleeding model. A possible mechanism for the preserved hemostasis was linked to platelet-fibrinogen interactions. Both Fga270/270 and siFga platelets readily aggregated initially, albeit both disaggregated faster following ADP stimulation. Fibrinogen-mediated platelet aggregation is dependent on the αIIbβ3 integrin, which was present at similar levels on the platelet membrane in all Fga270 mice. Additionally, αIIbβ3-dependent adhesion and spreading on immobilized FibAαWT and FibAα270 were similar.31 Unexpectedly, fibrinogen uptake in Fga270/270 platelets exceeded the anticipated amounts based solely on plasma levels. Integrin αIIbβ3 plays a major role in plasma protein uptake, but mechanisms that dictate the relative amounts of specific plasma factors within platelets remain largely undefined.32,33 Elevated fibrinogen levels in platelets relative to plasma levels was also observed in siFga-treated mice, indicating this is a phenomenon of hypofibrinogenemia and not linked specifically to the αC-region truncation. Intraplatelet fibronectin levels in Fga270/270 mice were significantly increased when compared with FgaWT/WT mice, in an apparent compensation for the reduced fibrinogen. This observation is similar to what has been described in Fga−/− mice and patients with afibrinogenemia.17,34 Moreover, platelets from mice expressing mutant fibrinogen lacking the binding site for integrin αIIbβ3 (FggΔ5/Δ5) have significantly reduced fibrinogen levels with elevated fibronectin levels.33 Given that the plasma fibrinogen levels of FggΔ5/Δ5 mice are normal, these findings suggest that intraplatelet fibronectin levels are regulated by and inversely correlated to intraplatelet fibrinogen levels.33
Truncation of the αC-region in FibAα270 resulted in profound changes in clot structure and stability. Turbidity analyses using purified fibrinogens indicated that the FibAα270 mutant protein produced clots with decreased optical density, consistent with studies using Fibrinogen Keokuk, Nieuwegein, Guarenas, Perth, Marburg, and Otago.2,3,7-10 In prior scanning electron microscope studies, recombinant human FibAα251 produced denser clots with thinner fibers.35 However, FibAα270 produced heterogenous clots that are generally less dense with thicker fibers, an observation similar to that for fibrinogen truncated at residue 220 of the α chain.36 The basis for the structural differences in clots with different fibrinogen αC-region truncation mutants remains unknown. The αC region of fibrinogen also binds to components of the plasminogen activation system, and fibrin lacking the αC region has been suggested to have reduced susceptibility to fibrinolysis.5 Consistent with these findings, tPA/plasmin-mediated fibrinolysis of FibAα270 clots and plasmin generation in Fga270/270 plasma did not occur in our in vitro analyses. Although previous studies suggested that reducing or eliminating plasmin(ogen) has little impact on hemostasis,37,38 it is possible that, in combination with the altered clot structure of FibAα270, the loss of fibrinolytic potential facilitates hemostasis. Consistently, fibrin accumulation progressively increased in the hemostatic plugs of Fga270/270 mice following successive saphenous vein laser injury despite low plasma fibrinogen, which could have contributed to the preservation of hemostatic potential.
Previous research indicated that hyperfibrinogenemia exacerbates thrombosis.39 A largely untested hypothesis is whether selectively lowering fibrinogen levels confers protection from thrombotic disease. We found no reports of thrombosis in αC-region truncation patients in the absence of interventions. There are reports of hypofibrinogenemic Fibrinogen Keokuk and Fibrinogen Marburg patients developing thrombosis, but only after cryoprecipitate or fresh blood transfusion.3,10 Animal models and in vitro systems each indicate that fibrin(ogen) supports the development of occlusive thrombi.12,16Fga−/− and mice expressing a mutant form of fibrinogen that cannot support fibrin formation (FgaAEK/AEK) fail to develop occlusive thrombi, but each exhibit profound bleeding.12 Our studies document for the first time that genetically or pharmacologically lowering fibrinogen levels (while leaving other coagulation system components unaltered) is protective against venous thrombosis, while preserving hemostatic potential. All Fga270/270 mice and 4/6 siFga-treated mice developed no quantifiable thrombi in the IVC stasis model. Notably, 2/6 siFga-treated mice, whose circulating fibrinogen levels were ∼0.6 mg/mL, developed thrombi, but had smaller thrombi compared with siLuc-treated mice. Thus, pharmacologically lowering fibrinogen levels is beneficial even if venous thrombosis does occur. These findings also suggest that the loss of the fibrinogen αC-region itself may independently contribute to the protection against venous thrombosis. FXIIIa-mediated crosslinking of fibrinogen α chain contributes to thrombus size by promoting red blood cell retention in clots.19,20Although Fga270/270 plasma supports FXIII activation equivalently to FgaWT/WT plasma, the primary FXIIIa crosslinking residues on the Aα-chain that reside within the αC region are deleted in FibAα270, resulting in inability to form fibrinogen α-chain high-molecular-weight polymers.19,20,30,40 Furthermore, the αC region of fibrinogen binds the glycoprotein VI receptor on the platelet surface, which also contributes to the pathogenesis of venous thrombosis.41,42 Thus, Fga270/270 mice seem to be protected from experimental venous thrombosis through multiple mechanisms.
Fibrinogen plays an essential role in antimicrobial host defense. In an S aureus peritoneal infection model, Fga−/− and FgaAEK/AEK mice displayed compromised survival and S aureus clearance within the peritoneal cavity, highlighting an essential role of fibrin(ogen) in antimicrobial mechanisms.12,43 Here, we show that both Fga270/270 and FgaWT/WT mice had similar bacterial clearance and survival following intraperitoneal S aureus infection. Furthermore, bacterial killing and suppression of dissemination were maintained in siFga-treated mice. Together, these findings highlight that neither a 90% reduction of circulating fibrinogen nor loss of the fibrinogen αC region compromise fibrin(ogen)-dependent host defense mechanisms against S aureus infection.
In conclusion, our study reveals that Fga270/270 mice are hypodysfibrinogenemic with preserved hemostasis and fibrin(ogen)-dependent antimicrobial function, while exhibiting near complete protection from venous thrombosis. The αC-region variants may have clinical significance in nonhemostatic functions, given the observations implicating the αC region in the pathogenesis of Alzheimer's disease and familial renal amyloidosis.44,45Fga270 mice establish a promising model to examine the functional significance of an αC-region truncation mutation and to investigate the impact of profound hypofibrinogenemia in vivo.
Acknowledgments
The authors thank Sara Abrahams for her technical assistance and Bas de Laat for his generous gift of the α2-macroglobulin-plasmin complex for the plasmin generation assays.
This work was supported by grants from the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Disease (R01DK112778 to M.J.F. and R01DK120289 and R01DK122813 to J.P.L.), the National Cancer Institute (R01CA211098 to M.J.F.), the National Heart, Lung, and Blood Institute (R35HL144976 to W.B. and R01HL126974 to A.S.W.), and Nanomedicines Innovation Network of the Networks of Centres of Excellence of Canada (2020-T2-02 to C.J.K.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: W.S.H., E.G.B., D.S.P., E.G.C., O.A.N., J.-M.M., L.G.P., H.M.C.-F., J.P.L., and M.J.F. designed the research, performed experiments, and analyzed the data; L.J.J., J.L., and C.J.K. provided valuable reagents; W.S.H. and M.J.F. wrote the manuscript; and T.P.U., A.S.W., W.B., and J.P.L. provided critical guidance on experimental procedures and helped write the manuscript. All authors read and approved the final manuscript.
Conflict-of-interest disclosure: C.J.K. is a director and shareholder of NanoVation Therapeutics, Inc., which is developing other RNA-based therapies. C.J.K., L.J.J., and J.L. are inventors on pending intellectual property related to RNA-based therapies. The remaining authors declare no competing financial interests.
Correspondence: Matthew J. Flick, Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, 8018B Mary Ellen Jones Building, Chapel Hill, NC 27599-7035; e-mail: matthew_flick@med.unc.edu.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal