

In this issue of Blood, Pindzola et al1 used mice carrying 4 hallmark genetic alterations associated with the MYD88/CD79B-mutated (MCD) genetic subgroup of diffuse large B-cell lymphoma (DLBCL) to identify splenic germinal center B cells (GCBs) as a likely cell of origin for MCD-DLBCL.
DLBCL is the most common and aggressive lymphoma and comprises a heterogeneous group of mature B-cell cancers with diverse genetic mutations and variable clinical outcomes.2 DLBCL is commonly divided into 2 distinct subgroups, activated B-cell–like (ABC) and GCB lymphomas, based on gene expression profiles.3 MCD-DLBCL is a genetic subset of ABC-DLBCL with gain-of-function mutations in both MYD88 and CD79B.4 DLBCL originates in germinal centers (GCs) that form in the B-cell follicles of secondary lymphoid organs in response to antigen challenge. As shown (see figure), GCs have 2 functionally distinct regions, including the dark zone (DZ) and the light zone (LZ). B cells undergo robust proliferation and somatic hypermutation in the DZ and affinity-driven selection in the LZ. Ultimately, GCBs give rise to memory B cells (MBC) and plasmablasts (PB)/plasma cells (PC). ABC-DLBCL originates from GCBs that are committed to becoming PB3; however, there is recent evidence that MCD-DLBCL instead derives from aberrant MBC.5 Theprecise cell type from which MCD-DLBCL originates is not known. It is also unclear precisely how genetic alterations evolve during MCD-DLBCL development.
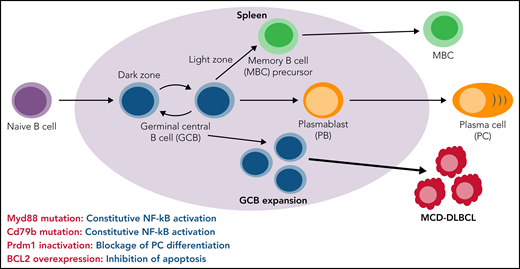
MYD88 and CD79B mutation-induced constitutive NF-kB activation, Prdm1 inactivation-caused blockade of PC differentiation, and BCL2 overexpression mediated inhibition of apoptosis collectively drive aberrant spontaneous expansion of splenic GCBs and ultimately result in the development of MCD-DLBCL in a mouse model.
MYD88 and CD79B mutation-induced constitutive NF-kB activation, Prdm1 inactivation-caused blockade of PC differentiation, and BCL2 overexpression mediated inhibition of apoptosis collectively drive aberrant spontaneous expansion of splenic GCBs and ultimately result in the development of MCD-DLBCL in a mouse model.
There is evidence that MYD88 and CD79B mutations are necessary but not sufficient for generation of MCD-DLBCL. MYD88 is an adaptor protein for the Toll/Interleukin-1 receptor. The MYD88 L265P mutation results in constitutive activation of the Toll-like receptor (TLR) and enhances B-cell receptor (BCR)–mediated activation of nuclear factor κB (NF-κB), thereby promoting B-cell proliferation and survival.6 Previous studies have shown that B-cell–specific expression of mutant MYD88 results in expansion of PC in vivo; however, there is controversy as to whether mutant MYD88 alone is sufficient to drive DLBCL in mice.7 The CD79A/CD79B heterodimer is a critical signaling component for initiating the BCR signaling cascade, which activates the NF-κB pathway and is essential for genesis of B-cell lymphomas. Mutation of CD79B occurs frequently in ABC-DLBCL, but the mutation is alone insufficient to activate NF-κB in ABC-DLBCL cells; instead, CD79B mutation increases cell-surface expression and clustering of the BCR, which leads to self-antigen–dependent chronic active BCR signaling and subsequent survival of ABC-DLBCL cells.2,8 Together, mutant MYD88 and CD79B enable creation of a My-T–BCR supercomplex consisting of MYD88, TLR9, and the immunoglobulin M (IgM) BCR, which drives constitutive NF-κB activation in the MCD subgroup and prevents apoptosis of lymphoma cells.6,9 Pindzola et al expanded upon these findings by showing that expression of mutant MYD88 in mature B cells in mice led to a cell-intrinsic spontaneous expansion of splenic GCBs and that mutant CD79B could synergize with mutant MYD88 to drive expansion of IgM+ splenic GCBs and accumulation of PC; however, mutant MYD88 alone or together with mutant CD79B was again insufficient to drive development of DLBCL in mice.
Collectively, these findings indicate that the contributions of mutant MYD88 and CD79B to MCD-DLBCL initiation are limited in the absence of other genetic lesions. Hence, progression to MCD-DLBCL from expanded GCBs must be preceded by the acquisition of additional genetic mutations. Pindzola et al explored the potential for additional genetic alterations to promote the generation of MCD-DLBCL. BCR-induced NF-κB activation increases the expression of IRF4, which induces the expression of the PC lineage-determining transcription factor PRDM1 (Blimp-1). PRDM1 and IRF4 together suppress BCL6 and drive GCB differentiation into PC. Loss of Prdm1 promotes DLBCL by blocking PC differentiation. Pindzola et al found that Prdm1 deletion and mutation of both MYD88 and CD79B in mice collaborated to result in spontaneous expansion of splenic GCBs with simultaneous increase in proliferation and apoptosis. Inhibiting cell apoptosis by overexpression of BCL2, in combination with MYD88/CD79B mutations and Prdm1 deletion, led to a massive spontaneous expansion of splenic GCBs in young mice and development of DLBCL in old mice. Taken together, these findings clearly demonstrate that the principle of co-occurring genetic alterations applies to development of MCD-DLBCL. Specifically, MYD88/CD79B mutation-induced constitutive NF-kB activation, Prdm-1 deletion/inactivation-induced blockade of PC differentiation, and BCL2 overexpression–mediated inhibition of apoptosis are collectively necessary for spontaneous expansion of aberrant splenic GCBs and ultimate development of MCD-DLBCL in a mouse model of human disease (see figure).
Fully understanding the interrelationships among the genetic alterations that give rise to MCD-DLBCL will provide new insights into disease pathogenesis and suggest new combination treatment strategies. Bulk and single-cell sequencing studies of the landscape of genetic mutations in patients with MCD-DLBCL and in elderly individuals predisposed to development of this disease will allow for identification of the stepwise order, and the B-cell developmental stages, in which genetic alterations in MYD88, CD79B, PRDM1, and BCL2 occur and give rise to generation of MCD-DLBCL in humans. It is possible that the chronological sequence of these cooperating genetic alterations and the B-cell developmental stage(s) in which these alterations arise vary considerably across patients and account for at least some of the substantial heterogeneity observed in human MCD-DLBCL. It is known that other genetic mutations, in addition to those examined by Pindzola et al, contribute to immune evasion and in similar or different ways promote development of MCD-DLBCL. Specific additional candidates include Pim1, genetic alteration of which is frequently associated with NF-kB activation, and TBL1XR, BCL6, or ETV6 alterations of which block PC differentiation.2 A limitation of the mouse model employed by Pindzola et al is that it is not able to determine the order in which the multiple genetic mutations must arise, nor the precise B-cell development stage(s) in which genetic alterations must occur, to trigger development and progression of MCD-DLBCL in human patients. Nevertheless, mouse models are valuable tools for identifying candidates that drive disease development and progression and for testing the efficacy of new combination therapies for aggressive MCD-DLBCL.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal