

TO THE EDITOR:
Richter syndrome (RS) is the histological transformation of chronic lymphocytic leukemia (CLL) into an aggressive lymphoma, typically diffuse large B-cell lymphoma (DLBCL), with most cases clonally related to the preceding CLL.1-3 In the past 10 years, a revolution has occurred in therapeutic options for patients with CLL.4 However, little has changed for patients with RS, with only a few phase 1 and 2 clinical trials ongoing, mostly based on different drug combinations.5 Novel therapeutic perspectives for RS may come from immune-based therapies6,7 or, alternatively, from the use of antibody-drug conjugates (ADCs) targeting antigens predominantly or exclusively expressed by neoplastic cells.8
One of these antigens is CD37, a heavily glycosylated leukocyte surface antigen belonging to the tetraspanin superfamily, selectively expressed at high levels by normal mature B cells, as well by mature B-cell malignancies, including non-Hodgkin lymphoma (NHL), CLL, and DLBCL.9-11 CD37 has recently gained attention as a potential target, with consequent development of novel therapeutic agents12-16 that have shown clinical efficacy in CLL and DLBCL when used in combination with rituximab, chemotherapy, or targeted compounds.17-21
The increased interest and efficacy of anti-CD37–based therapies prompted the development of 3 different anti-CD37-ATACs (amanitin-based ADCs). These novel drugs are composed of a chimeric monoclonal IgG1 antibody specific for CD37, conjugated via either a proteolytic cleavable (anti-CD37-Ama 1 and anti-CD37-Ama 2) or a noncleavable (anti-CD37-Ama 3) linker to amanitin as the payload (supplemental Figure 1A, available on the Blood Web site).
All 3 anti-CD37 ATACs showed full-blown cytotoxicity on target-positive Raji cells (Burkitt lymphoma cell line) with half-maximal effective concentration in the low nanomolar and picomolar range (anti-CD37-Ama 1: 2.04 × 10−9 M; anti-CD37-Ama 2: 7.5 × 10−10 M; anti-CD37-Ama 3: 3.9 × 10−9 M; supplemental Figure 1B). Target specificity was demonstrated by absence of cytotoxicity up to a concentration of 10−6 M in a CD37− cell line (HEK293wt; supplemental Figure 1C). In contrast, free α-amanitin showed cytotoxicity on Raji and HEK cells (supplemental Figure 1D) with half-maximal effective concentration values in the micromolar range, which was even lower in HEK cells expressing OATP1B3 (supplemental Figure 1D), a cell surface transporter that is known to transport α-amanitin into hepatocytes.
Amanitin is an RNA polymerase II inhibitor that interrupts cellular transcription at low nanomolar concentrations.22 Consistently, anti-CD37 ATACs induced cytotoxicity by arresting RNA transcription, with the active metabolite of anti-CD37-Ama 2 displaying a similar degree of inhibition of free α-amanitin, with a half-maximal inhibitory concentration in the low nanomolar range (supplemental Figure 1E).
Analysis of CD37 expression in cell lines and primary samples obtained from B-cell hematological malignancies revealed low levels of expression in acute lymphoblastic leukemia, Hodgkin lymphoma, and multiple myeloma, whereas CLL, mantle, and NHL cells expressed it at high levels (supplemental Figure 2A). Primary samples confirmed this pattern of expression, with CLL cells showing the highest expression, followed by follicular lymphoma and DLBCL, whereas acute lymphoblastic leukemia and multiple myeloma had low-to-undetectable expression of CD37 (supplemental Figure 2B). Importantly, RNA sequencing data performed on 14 patients with RS showed that CD37 was invariably expressed by this lymphoma (supplemental Figure 2B), as confirmed by immunohistochemical staining performed on bone marrow or lymph node biopsy specimens from patients with RS (Figure 1A). In paired CLL-RS specimens, CD37 expression, which was high in the CLL phase, was invariably maintained after RS transformation (Figure 1A).
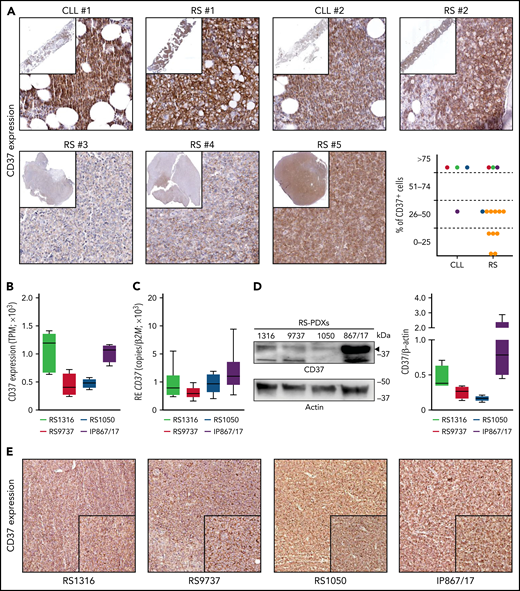
CD37 is expressed by Richter syndrome primary samples and patient-derived xenograft models. CD37 expression on primary RS samples was checked by immunohistochemistry performed on bone marrow or lymph node biopsy specimens from patients with RS (yellow dots) or paired CLL-RS samples (A; red-green-dark blue-purple dots). Representative staining of 2 paired CLL-RS and 3 RS samples. Original magnification, ×200; inset ×6. CD37 expression was checked, both at the transcript and protein levels, in our 4 RS-PDX models (RS1316, RS9737, RS1050, and IP867/17) by RNA sequencing (B; n = 4 per model; box and whiskers, minimum to maximum representation), quantitative real-time PCR (C; n = 8 per model; box and whiskers, minimum to maximum representation), western blot analysis (D; n = 4 per model; box and whiskers, minimum to maximum representation) and immunohistochemistry staining on slides of formalin-fixed and paraffin-embedded, RS-PDX–derived tumor masses (E). In western blot analyses, actin was used as a loading control. RE, relative expression; TPM, transcript per million.
CD37 is expressed by Richter syndrome primary samples and patient-derived xenograft models. CD37 expression on primary RS samples was checked by immunohistochemistry performed on bone marrow or lymph node biopsy specimens from patients with RS (yellow dots) or paired CLL-RS samples (A; red-green-dark blue-purple dots). Representative staining of 2 paired CLL-RS and 3 RS samples. Original magnification, ×200; inset ×6. CD37 expression was checked, both at the transcript and protein levels, in our 4 RS-PDX models (RS1316, RS9737, RS1050, and IP867/17) by RNA sequencing (B; n = 4 per model; box and whiskers, minimum to maximum representation), quantitative real-time PCR (C; n = 8 per model; box and whiskers, minimum to maximum representation), western blot analysis (D; n = 4 per model; box and whiskers, minimum to maximum representation) and immunohistochemistry staining on slides of formalin-fixed and paraffin-embedded, RS-PDX–derived tumor masses (E). In western blot analyses, actin was used as a loading control. RE, relative expression; TPM, transcript per million.
We then took advantage of established PDX models, which have been shown to be useful tools to test the efficacy of novel drugs/drug combinations.8,23,24 The 4 RS-PDX models were characterized by significant levels of CD37, with 2 of them, RS1316 and IP867/17, showing slightly higher CD37 expression compared with the other 2 (RS9737 and RS1050; Figure 1B-D). Flow cytometry analyses (supplemental Figure 2C) and immunohistochemistry staining on formalin-fixed, paraffin-embedded sections of tumor masses obtained from the 4 RS-PDXs (Figure 1E) confirmed these observations.
All 3 anti-CD37 ATACs (anti-CD37-Ama 1, -Ama 2, and -Ama 3) bound CD37 (supplemental Figure 2D), ruling out any interference of the linker or amatoxin with antibody-binding properties. When tested in vitro (40 and 200 nM; 72 hours) on RS-PDX–derived cells, these 3 ATACs resulted in a significant apoptotic response, with limited heterogeneity among the PDXs and compounds (supplemental Figure 3B). These results were corroborated by pronounced cleavage of caspase-3 and PARP proteins in all models (supplemental Figure 3B). All anti-CD37 ATACs induced apoptosis selectively in CD37-expressing cells. Indeed, when incubated with peripheral blood mononuclear cells from healthy donors, only CD37+ B cells underwent apoptosis, whereas no reduction in viability was measured in T lymphocytes (CD37−; supplemental Figure 4). Furthermore, no toxicity was assessed when treating these cells with nontargeted antibodies conjugated to amanitin with the same 3 linkers, underlining the specificity of anti-CD37 ATACs (supplemental Figure 4).
Finally, these ATACs were tested in vivo in 3 of 4 RS-PDX models (RS1316, RS9737, and RS1050), which can grow in a systemic way, mimicking the human disease. After IV injection, RS cells are engrafted and localized in different tissues, including spleen, bone marrow, peripheral blood, and even the brain, effectively establishing a progressive lymphoma.25 Anti-CD37 ATACs were administered IV at 2 different doses as a single treatment (anti-CD37-Ama 1: 2.5 and 5 mg/kg; anti-CD37-Ama 2: 5 and 10 mg/kg; and anti-CD37-Ama 3: 20 and 40 mg/kg). The mice were then compared with a vehicle control group for survival and disease distribution in different organs (spleen, liver, peripheral blood, bone marrow, kidney, brain, and lung), as assessed by flow cytometry using tumor-specific human markers. Consistent with the in vitro data, RS1316 showed the best responses in terms of improvement of survival after anti-CD37 ATAC administration, with complete disease regression in 21 of 24 treated mice (P = .0067; Figure 2A, left panel) and no or very limited tumor cells in the different organs (supplemental Figure 5A). At variance with RS1316, all RS9737, and RS1050 mice died of disease (supplemental Figure 5B), but anti-CD37 ATAC-treated mice displayed a marked increase in survival (Figure 2B). Specifically, mice treated with anti-CD37 ATACs displayed heterogenous responses among the 3 different compounds, with no significant differences between the 2 doses (vehicle: 24 days vs anti-CD37-Ama 2: 45 days vs anti-CD37-Ama 1: 61 days; P = .0091). Anti-CD37-Ama 3 showed a higher degree of heterogeneity of in vivo response (41-59 days with the higher dose vs 42-86 days with the lower one; P = .0091). Likewise, the RS1050 model showed a significant increase in survival when mice were treated with anti-CD37 ATACs, although with some differences among the compounds, and with the only exception of the anti-CD37-Ama 1 that resulted only in a mild in vivo effect (Figure 2B; right panel). Vehicle-treated mice were euthanized on days 32 to 34 after injection of RS cells, with neoplastic cells localizing in several tissues (supplemental Figure 5C). Anti-CD37-Ama 3 resulted in a statistically significant increased survival (41-69 days after injection), with comparable results when considering the 2 doses of drug (P = .0107 for the 40-mg/kg dose and P = .0193 for the lower dose). Anti-CD37-Ama 2 treatment further improved the mice’s survival (63-84 days for the higher dose and 65-120 days for the 5-mg/kg dose).
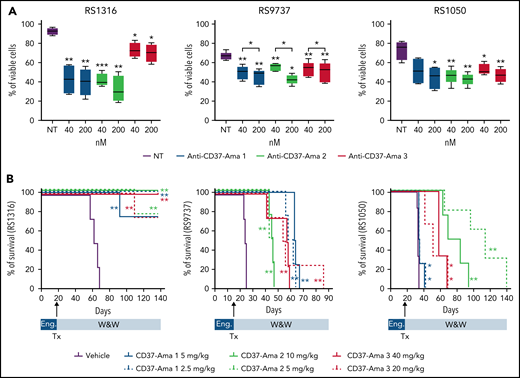
Anti-CD37 ATACs induce apoptosis in vitro and prolong survival of RS-PDX mice. (A) The apoptotic response of RS cells exposed for 72 hours to different doses (40 and 200 nM) of anti-CD37-Ama 1, anti-CD37-Ama 2, and anti-CD37-Ama 3 or left untreated was analyzed with conventional cytofluorimetric annexin V/PI staining (n = 5 per model; box and whiskers, minimum to maximum representation). (B) RS cells were injected IV into NOD/SCID/γ chain−/− mice and left to engraft (Eng) for 2 weeks. The mice were then randomly assigned to different treatment groups (4 mice per group). After anti-CD37 ATAC administration (Tx), mice were monitored (W&W, watch and wait) and survival analyses performed, generating Kaplan-Mayer curves. (A) Asterisks on the box plots refer to statistical significance vs the untreated condition; P values with bars refer to the indicated conditions. Statistical analyses were performed with the paired t test. *P < .05; **P < .01; ***P < .001.
Anti-CD37 ATACs induce apoptosis in vitro and prolong survival of RS-PDX mice. (A) The apoptotic response of RS cells exposed for 72 hours to different doses (40 and 200 nM) of anti-CD37-Ama 1, anti-CD37-Ama 2, and anti-CD37-Ama 3 or left untreated was analyzed with conventional cytofluorimetric annexin V/PI staining (n = 5 per model; box and whiskers, minimum to maximum representation). (B) RS cells were injected IV into NOD/SCID/γ chain−/− mice and left to engraft (Eng) for 2 weeks. The mice were then randomly assigned to different treatment groups (4 mice per group). After anti-CD37 ATAC administration (Tx), mice were monitored (W&W, watch and wait) and survival analyses performed, generating Kaplan-Mayer curves. (A) Asterisks on the box plots refer to statistical significance vs the untreated condition; P values with bars refer to the indicated conditions. Statistical analyses were performed with the paired t test. *P < .05; **P < .01; ***P < .001.
To exclude any toxicity related to amanitin administration, ad hoc experiments were performed on NSG mice to analyze the effects of this toxin on the liver, administering the 3 ADCs at the higher dose with the same schedule adopted for RS models. Hepatic enzymes were measured, and histology of the liver was performed at days 7 and 21 after administration of ADC. Elevation of aspartate aminotransferase and alanine aminotransferase was noted 7 days after treatment, with levels diminishing after 21 days. However, regarding hepatic tissue and morphology, no signs of hepatic toxicity related to the administration of amanitin was note at the histological level in treated mice, suggesting a transient elevation of the enzymes, but no permanent liver damage (supplemental Figure 6 and supplemental Table 2).
Taken together, the data indicate that CD37 may represent a good candidate for targeting RS cells with highly selective amanitin-based ADCs.
Acknowledgments
This work was supported by the Italian Association for Cancer Research (AIRC) (My First AIRC Grant MFAG-23107 (T.V.); investigator grant IG-23095 (S.D.); the Italian Ministry of Health (GR-2016-02364298 (T.V.); the Ministry of Education, University and Research-MIUR “Progetto strategico di Eccellenza Dipartimentale” grant D15D18000410001 (S.D.) as part of the Department of Medical Sciences, University of Turin; Fondazione CRT Erogazioni Ordinarie 2021 I tornata (T.V.); and Heidelberg Pharma research funds (S.D.).
Authorship
Contribution: T.V., C.O., M.K., A.P., and S.D. designed the research; M.G.P., O.J., and A.D. provided material; T.V., N.V., M.M., L.B., A.I., G.L., J.C.C., and A.D. performed the experiments; T.V., C.O., M.K., A.P., and S.D. analyzed the results, produced the figures, and wrote the manuscript; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: T.V. has received research funds from AstraZeneca. N.V., C.O., M.K., and A.P. are employed by Heidelberg Pharma. S.D. has received research funds from AstraZeneca, VelosBio Inc, and Heidelberg Pharma. The remaining authors declare no competing financial interests.
Correspondence: Silvia Deaglio, Department of Medical Sciences, University of Turin, Via Nizza 52, 10126 Torino, Turin, Italy; e-mail: silvia.deaglio@unito.it; and Tiziana Vaisitti, Department of Medical Sciences, University of Turin, Via Nizza 52, 10126 Torino, Turin, Italy; e-mail: tiziana.vaisitti@unito.it.
Original data are available by e-mail request to the corresponding author (silvia.deaglio@unito.it or tiziana.vaisitti@unito.it).
The online version of this article contains a data supplement.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal