

Key Points
Vamifeport reduced hemoglobin concentration in RBCs and diminished intravascular hemolysis and inflammation in a mouse model of SCD.
As a result, vamifeport improved hemodynamics and prevented vascular stasis in a mouse model of SCD.

Abstract
Sickle cell disease (SCD) is an inherited hemolytic anemia caused by a single point mutation in the β-globin gene of hemoglobin that leads to synthesis of sickle hemoglobin (HbS) in red blood cells (RBCs). HbS polymerizes in hypoxic conditions, leading to intravascular hemolysis, release of free hemoglobin and heme, and increased adhesion of blood cells to the endothelial vasculature, which causes painful vaso-occlusion and organ damage. HbS polymerization kinetics are strongly dependent on the intracellular HbS concentration; a relatively small reduction in cellular HbS concentration may prevent HbS polymerization and its sequelae. We hypothesized that iron restriction via blocking ferroportin, the unique iron transporter in mammals, might reduce HbS concentration in RBCs, thereby decreasing hemolysis, improving blood flow, and preventing vaso-occlusive events. Indeed, vamifeport (also known as VIT-2763), a clinical-stage oral ferroportin inhibitor, reduced hemolysis markers in the Townes model of SCD. The RBC indices of vamifeport-treated male and female Townes mice exhibited changes attributable to iron-restricted erythropoiesis: decreased corpuscular hemoglobin concentration mean and mean corpuscular volume, as well as increased hypochromic and microcytic RBC fractions. Furthermore, vamifeport reduced plasma soluble VCAM-1 concentrations, which suggests lowered vascular inflammation. Accordingly, intravital video microscopy of fluorescently labeled blood cells in the microvasculature of Townes mice treated with vamifeport revealed diminished adhesion to the endothelium and improved hemodynamics. These preclinical data provide a strong proof-of-concept for vamifeport in the Townes model of SCD and support further development of this compound as a potential novel therapy in SCD.
Introduction
Sickle cell disease (SCD) is an autosomal recessive hemolytic disorder caused by a single nucleotide substitution in the β-globin gene that results in the synthesis of sickle hemoglobin (HbS) and sickle-shaped red blood cells (RBCs).1,2 Upon deoxygenation in capillaries, HbS polymerizes in rigid fibers, causing RBC membrane distortion that may lead to intravascular hemolysis and inflammation. Intravascular hemolysis is a major pathophysiological mechanism in SCD resulting in the release of cell-free hemoglobin into plasma, which oxidizes and liberates reactive heme into the vasculature. Cell-free heme activates endothelial cell adhesion molecules and induces leukocyte activation and migration, as well as release of reactive oxygen species, cytokines, and chemokines, that all together may lead to vaso-occlusion and pain crisis.3-6 Extravascular hemolysis is also prominent in SCD, resulting from the removal of damaged RBCs by macrophages.7,8
The time before HbS forms fibers (“delay time of gelation”) is exponentially proportional to intracellular HbS concentration.9,10 Even a HbS decrease of ∼5% to 7% may increase the delay time of gelation sufficiently to allow RBCs to cross capillaries without sickling.11,12 One possible approach to lower RBC HbS concentration is by inducing iron-restricted erythropoiesis, as iron is necessary for hemoglobin synthesis.13 Iron deficiency in patients with SCD decreases mean corpuscular HbS concentration, intravascular hemolysis, and pain crisis.14-16 One retrospective study showed clinical improvements in 71% of patients with hemoglobin SC disease undergoing regular phlebotomies.17 Conversely, higher iron availability was associated with nocturnal hypoxia in children with SCD.18
The concept of therapeutic iron restriction in SCD has not been tested in large controlled trials, mostly due to difficulties in inducing an appropriate level of iron deficiency without rendering patients severely anemic. In mouse models, iron deficiency induced by either a low-iron diet (LID) or genetic disruption of intestinal hypoxia-inducible factor-2α (HIF-2α) improved anemia and hemolytic markers,19,20 mechanistically supporting the therapeutic potential of iron limitation.
We hypothesized that iron restriction by blocking ferroportin, the unique iron transporter in mammals,21 may lower HbS concentrations in RBCs, thereby reducing hemolysis, improving blood flow, and preventing vaso-occlusive events. Vamifeport (VIT-2763) is a small-molecule ferroportin inhibitor that, like hepcidin, binds to ferroportin, thereby blocking dietary iron absorption and iron export from liver and spleen macrophages.22 We therefore tested the efficacy of vamifeport in the Townes mouse model of SCD.
Methods
Animal model
The knock-in Townes model was developed by replacing the mouse α- and β-globin genes with both human α-globin and β-globin (human γA and βA or sickle βS form) genes.23 Mice homozygous for the sickle βS form are referred to as HbSS mice; they are characterized by hemolytic anemia, high reticulocyte counts, and splenomegaly, and mimic many pathophysiological features of severe SCD in humans.23 Mice homozygous for human βA are referred to as HbAA mice, are phenotypically normal, and were used as controls. Mice were purchased from The Jackson Laboratory (JAX stock #013071; Bar Harbor, ME) and bred under pathogen-free conditions in the Vifor (International) Ltd. animal facility (Schlieren, Switzerland) according to Swiss veterinary law. Animals were group-housed (3-5 mice per cage) under a 12-hour reverse dark/light cycle and provided with nesting material, enrichment materials, and water/food ad libitum (standard rodent diet; 250 ppm iron content, unless otherwise stated; Granovit SA, Kaiseraugst, Switzerland).
Ethics statement
Animal experiments complied with Swiss law and associated guidelines for animal experimentation and were approved by the responsible authority (Veterinary Department, Canton Zurich; permit number ZH108/2017). Studies were performed in compliance with the Vifor Pharma Group Code of Conduct and comply with the principles of 3R (Replace, Reduce, Refine). Animals were tunnel handled, had a tunnel in their cage for shelter, and were scored daily for body weight and physical condition.
Experimental design and treatment with vamifeport
HbSS mice were randomly distributed to cages at weaning and allocated to treatment groups according to a stratified randomization. Six- to seven-week-old HbSS mice (n = 8-12) were treated with either vehicle (0.5% methylcellulose) or vamifeport 60 mg/kg (5 mL/kg) twice daily (6 hours apart) by oral gavage for 6 weeks. Age-matched male and female HbAA mice (n = 8-10) dosed with vehicle served as controls. Dosing was performed at the start of the animals’ active phase (facility dark phase). To distinguish preexisting iron from that absorbed during the study, all mice were fed LID (<10 mg/kg iron; Granovit SA) during the full study period, and the stable iron isotope 58Fe was supplemented in the drinking water (1 mM 58FeSO4 with 10 mM ascorbic acid as reducing agent, corresponding to iron uptake in standard rodent diet of ∼250 ppm iron) for 6 hours after the first daily dose of compound/vehicle. For the remaining 18 hours after the second dose, all animals were given mineral water without iron. Hemoglobin concentrations were determined weekly in tail vein blood (HemoCue AB, Ängelholm, Sweden). Animals were euthanized by complete exsanguination after terminal anesthesia with isoflurane 3 hours after final dosing.
To study the effects of pharmacologic vs dietary iron restriction, 6- to 7-week-old HbSS mice (n = 8-12) were either fed LID with 58Fe-supplemented water and treated with vamifeport as described in the preceding paragraph or they were fed an LID without iron supplementation.
For assessment of hemodynamics and vaso-occlusion by intravital microscopy, 6- to 12-week-old HbSS mice (n = 8) were treated with vamifeport 30 mg/kg or 100 mg/kg orally twice daily for 4 weeks. HbSS and vehicle-treated HbAA mice served as controls.
Oxygen gradient ektacytometry (oxygenscan)
Blood was obtained by tail vein puncture and stored at 4°C up to 24 hours before analysis. Deformability of RBCs expressed as the elongation index (EI) was measured as a function of oxygen pressure using the oxygenscan test on a laser optical red cell rotational analyzer (Lorrca, RR Mechatronics, Zwaag, The Netherlands). Oxygenscan measurements were performed with 400 × 106 RBCs for all samples and according to the manufacturer’s protocol and standard parameters.
Intravital microscopy analysis of vaso-occlusion
At day 28, after 4 weeks of vamifeport treatment, dorsal skin-fold window chamber surgery was performed on anesthetized mice, as described previously.24 Hemodynamics and blood cell adhesion to microvasculature were investigated by using intravital microscopy 90 minutes after induction of vaso-occlusion by murine recombinant tumor necrosis factor-α (500 ng/mouse).25 Videos were recorded at different locations within the dorsal skin-fold window chamber to determine the effect of vamifeport on blood cell adhesion to the vascular endothelium and vaso-occlusion. At least 8 venules per mouse were recorded for a total of ≥60 minutes. Videos were produced by using 10× and 20× magnifications. Cell adhesion was quantified by measuring the fluorescence intensity of adherent fluorescence-labeled cells on still images (20× magnification) using ImageJ software (Version: 2.3.0/1.53q downloaded from the NIH website).26 Blood flow was determined by counting the number of vessels with normal blood flow, slow blood flow, and no blood flow according to frame-by-frame analysis of video replay. Values obtained from analyzing all recorded vessel segments were averaged across groups (n = 4-7 mice per group).
Statistical analysis
A priori sample size calculations were performed with estimated effect size for several parameters from pilot experiments, target power of 0.8, and correction of α for multiple comparisons using G*Power 3.1.9.7.27 Parameters over time were analyzed by using two-way analysis of variance with repeated measures for time-course effects. Where significant effects were observed, post hoc tests were performed by using Dunnett’s adjustment for multiple comparisons, where each group was compared with HbSS vehicle controls. End point parameters were analyzed by using one-way analysis of variance with Dunnett’s multiple comparison test. Data are presented by individual value with mean as scatter plots. Analyses were conducted by using GraphPad Prism version 8.4.2 (GraphPad Software, San Diego, CA).
Results
Vamifeport induced iron-restricted erythropoiesis with reduced hemoglobin concentration in RBCs
Vamifeport induced iron-restricted erythropoiesis in HbSS mice, as shown by a 5% reduction of the corpuscular hemoglobin concentration mean (CHCM) (Figure 1A) compared with the HbSS vehicle group (CHCM is a direct measure of hemoglobin concentrations within intact RBCs). Furthermore, vamifeport decreased mean corpuscular hemoglobin content, mean cellular volume (MCV), and hematocrit (likely as a function of lowered MCV), without affecting RBC counts, or durably decreasing the total blood hemoglobin concentration, suggesting that at the doses tested, vamifeport did not importantly exacerbate anemia.
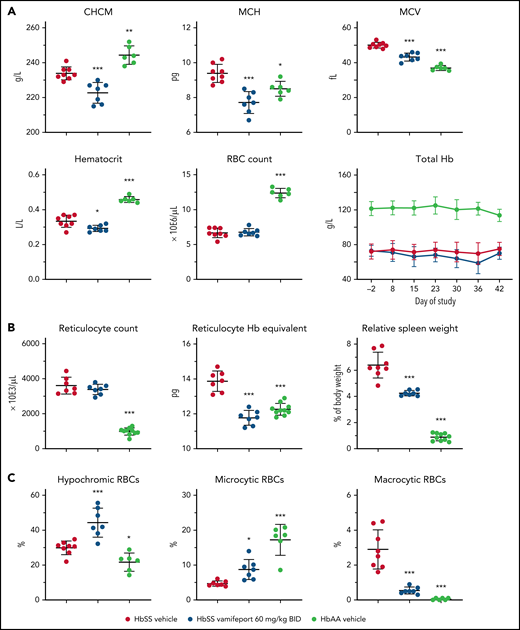
Vamifeport induced iron-restricted erythropoiesis with reduced hemoglobin (Hb) concentration in RBCs of HbSS mice. (A) Administration of vamifeport (60 mg/kg) twice daily (BID) for 6 weeks in 6- to 7-week-old HbSS mice resulted in iron-restricted erythropoiesis, as shown by decreased CHCM, mean corpuscular hemoglobin (MCH), MCV, hematocrit, and total Hb without lowering RBC counts. (B) Likewise, reticulocyte counts remained stable with marked reductions in reticulocyte Hb and spleen size. (C) RBC scatterplots revealed a significant increase in the proportion of hypochromic and microcytic RBCs after vamifeport treatment. Results are presented as individual values with mean ± standard deviation (n = 6-8 mice per group). Analysis was performed by using one-way analysis of variance with Dunnett’s multiple comparison of all groups vs the HbSS vehicle group: *P < .05, **P < .01, ***P < .001.
Vamifeport induced iron-restricted erythropoiesis with reduced hemoglobin (Hb) concentration in RBCs of HbSS mice. (A) Administration of vamifeport (60 mg/kg) twice daily (BID) for 6 weeks in 6- to 7-week-old HbSS mice resulted in iron-restricted erythropoiesis, as shown by decreased CHCM, mean corpuscular hemoglobin (MCH), MCV, hematocrit, and total Hb without lowering RBC counts. (B) Likewise, reticulocyte counts remained stable with marked reductions in reticulocyte Hb and spleen size. (C) RBC scatterplots revealed a significant increase in the proportion of hypochromic and microcytic RBCs after vamifeport treatment. Results are presented as individual values with mean ± standard deviation (n = 6-8 mice per group). Analysis was performed by using one-way analysis of variance with Dunnett’s multiple comparison of all groups vs the HbSS vehicle group: *P < .05, **P < .01, ***P < .001.
Reticulocyte counts are increased in SCD in response to chronic hypoxia, which stimulates stress erythropoiesis in bone marrow and spleen, leading to release of immature RBCs.19 Vamifeport did not change absolute reticulocyte counts in HbSS mice; however, it significantly lowered reticulocyte hemoglobin content (Figure 1B). In SCD mice, hemolytic anemia and hypoxia trigger compensatory extramedullary erythropoiesis in the spleen, leading to expanded red pulp and splenomegaly.23 Vamifeport reduced the spleen weight of HbSS mice by 31% compared with vehicle-treated HbSS control mice, indicating amelioration of the extramedullary erythropoiesis. Consistent with inducing iron-restricted erythropoiesis, vamifeport significantly increased the proportion of hypochromic and microcytic RBCs, while reducing the percentage of macrocytic RBCs (Figure 1C).
Vamifeport reduced hemolysis
Plasma markers of hemolysis, including total heme, lactate dehydrogenase, and indirect bilirubin, were reduced in vamifeport-treated HbSS mice, suggesting that iron-restricted erythropoiesis induced by ferroportin inhibition lowers intravascular and extravascular hemolysis (Figure 2A-C). Vamifeport decreased the proportion of macrophages that had taken up RBCs (Figure 2D), further indicating that iron restriction by vamifeport attenuates extravascular hemolysis. Plasma hemoglobin and heme are filtered by the kidney, exposing the kidney to their associated adverse effects, and resulting in the accumulation of iron in proximal tubule epithelial cells.28,29 HbSS mice exhibited increased kidney iron concentrations compared with control mice, and vamifeport reduced kidney iron, indicating the potential of vamifeport in preventing iron-mediated kidney injury (Figure 2E). Perls staining of kidney sections from vehicle-treated HbSS mice confirmed intense tubular iron accumulation, which was absent in HbAA mice and strongly diminished in vamifeport-treated HbSS mice (Figure 2F).
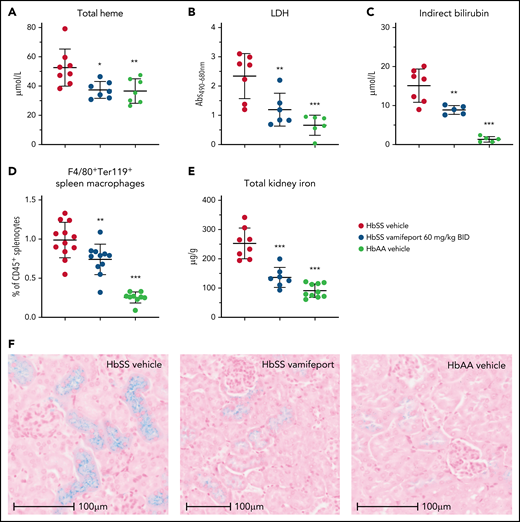
Vamifeport reduced hemolysis in HbSS mice. Vamifeport significantly reduced plasma markers of hemolysis in HbSS mice, including heme (A), lactate dehydrogenase (LDH) (B), and indirect bilirubin (C). (D) Erythrophagocytosis was reduced in HbSS mice treated with vamifeport, as shown by percentages of Ter119+ red pulp macrophages, identified by gating on CD45+F4/80+ spleen cells. (E) Total iron deposition in the kidneys of HbSS mice was reduced by vamifeport, as measured by inductively coupled plasma optical emission spectrometry. (F) Reduction of total iron deposition in the kidneys of HbSS mice by vamifeport was also illustrated by Perls staining of kidney sections. Results are presented as individual values with mean ± standard deviation (n = 5-8 mice per group). Analysis was performed by using one-way analysis of variance with Dunnett’s multiple comparison of all groups vs the HbSS vehicle group: *P < .05, **P < .01, ***P < .001. Abs, absorbance; BID, twice daily.
Vamifeport reduced hemolysis in HbSS mice. Vamifeport significantly reduced plasma markers of hemolysis in HbSS mice, including heme (A), lactate dehydrogenase (LDH) (B), and indirect bilirubin (C). (D) Erythrophagocytosis was reduced in HbSS mice treated with vamifeport, as shown by percentages of Ter119+ red pulp macrophages, identified by gating on CD45+F4/80+ spleen cells. (E) Total iron deposition in the kidneys of HbSS mice was reduced by vamifeport, as measured by inductively coupled plasma optical emission spectrometry. (F) Reduction of total iron deposition in the kidneys of HbSS mice by vamifeport was also illustrated by Perls staining of kidney sections. Results are presented as individual values with mean ± standard deviation (n = 5-8 mice per group). Analysis was performed by using one-way analysis of variance with Dunnett’s multiple comparison of all groups vs the HbSS vehicle group: *P < .05, **P < .01, ***P < .001. Abs, absorbance; BID, twice daily.
Vamifeport improved RBC membrane composition and mitochondria clearance
To investigate the potential mechanism of how iron restriction by vamifeport reduces hemolysis, we studied the functional characteristics of RBCs. Phosphatidylserine (PS) exposure is increased on RBCs from patients with SCD.30,31 Externalized PS is prothrombotic and provides a potential adhesion site for phagocytes and activated endothelial cells, thus contributing to vascular dysfunction in SCD.32-34 Higher PS concentrations were observed on circulating RBCs from HbSS mice compared with HbAA mice (Figure 3A). Vamifeport significantly decreased the percentage and intensity of PS staining using Annexin V on circulating RBCs in HbSS mice, indicating that iron restriction by ferroportin inhibition diminishes eryptosis.
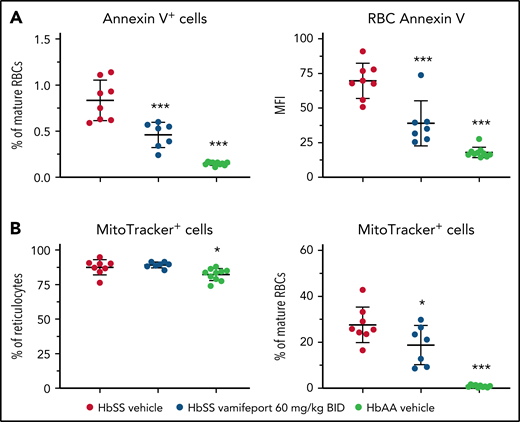
Vamifeport decreased PS exposure and improved mitochondria clearance in mature RBCs of HbSS mice. (A) Vamifeport lowered the exposure of PS on peripheral blood RBCs, as detected by a decreased percentage of Annexin V–positive cells and decreased intensity of Annexin V staining. (B) Although almost all reticulocytes of vehicle- and vamifeport-treated HbSS mice contained mitochondria (left), treatment with vamifeport reduced the occurrence of mitochondria in mature RBCs (right). Results are presented as individual values with mean ± standard deviation (n = 7-10 mice per group). Analysis was performed by using one-way analysis of variance with Dunnett’s multiple comparison of all groups vs the HbSS vehicle group: *P < .05, ***P < .001. BID, twice daily; MFI, mean fluorescent intensity.
Vamifeport decreased PS exposure and improved mitochondria clearance in mature RBCs of HbSS mice. (A) Vamifeport lowered the exposure of PS on peripheral blood RBCs, as detected by a decreased percentage of Annexin V–positive cells and decreased intensity of Annexin V staining. (B) Although almost all reticulocytes of vehicle- and vamifeport-treated HbSS mice contained mitochondria (left), treatment with vamifeport reduced the occurrence of mitochondria in mature RBCs (right). Results are presented as individual values with mean ± standard deviation (n = 7-10 mice per group). Analysis was performed by using one-way analysis of variance with Dunnett’s multiple comparison of all groups vs the HbSS vehicle group: *P < .05, ***P < .001. BID, twice daily; MFI, mean fluorescent intensity.
More than 80% of reticulocytes from both HbSS and HbAA mice contained mitochondria (Figure 3B). Upon maturation, 0.9% ± 0.5% of terminally differentiated RBCs retained mitochondria in HbAA control mice, compared with 27.6% ± 7.8% of mature RBCs in vehicle-treated HbSS mice. Vamifeport significantly reduced the proportion of mitochondria-containing mature RBCs in HbSS mice to 18.8% ± 8.5%. Because the remaining mitochondria in mature RBCs are a potential source of oxygen radicals, the improved clearance of mitochondria by vamifeport may decrease oxidative stress–induced hemolysis and eryptosis in SCD (Figures 2 and 3A).
Vamifeport reduced systemic and vascular inflammation, and oxidative stress
Townes mice and patients with SCD have elevated numbers of circulating leukocytes that produce proinflammatory cytokines and chemokines, thereby attracting further inflammatory cells and activating the endothelium.35,36 Analysis of peripheral blood leukocytes revealed amelioration of leukocytosis and neutrophilia in vamifeport-treated HbSS mice compared with vehicle-treated control mice (Figure 4A). Plasma concentrations of RANTES, which recruits leukocytes to proinflammatory sites, were significantly reduced in vamifeport-treated HbSS mice compared with vehicle-treated control mice (Figure 4B). The chemokine Cxcl1 expression was significantly upregulated in the livers of vehicle-treated HbSS mice compared with HbAA mice (Figure 4C). Vamifeport significantly reduced liver Cxcl1 expression in HbSS mice.
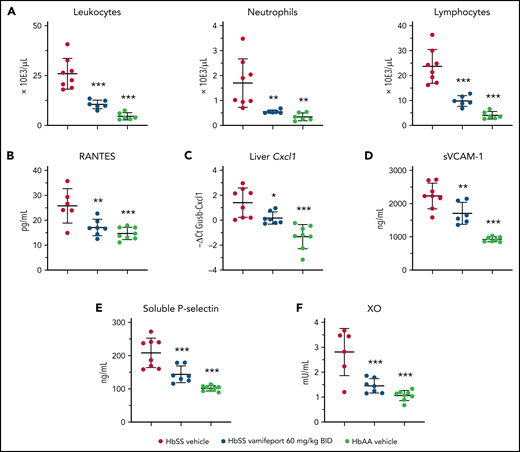
Vamifeport reduced systemic and vascular inflammation, and oxidative stress, in HbSS mice. Vamifeport significantly reduced inflammatory markers in HbSS mice: total leukocyte, neutrophil, and lymphocyte counts (A); plasma levels of the chemokine RANTES (B); expression of the neutrophil chemoattractant Cxcl1, as determined by quantitative polymerase chain reaction in liver (C); sVCAM-1 (D); soluble P-selectin (E); and plasma XO activity (F). Results are presented as individual values with mean ± standard deviation (n = 6-8 mice per group). Analysis was performed by using one-way analysis of variance with Dunnett’s multiple comparison of all groups vs the HbSS vehicle group: *P < .05, **P < .01, ***P < .001. BID, twice daily.
Vamifeport reduced systemic and vascular inflammation, and oxidative stress, in HbSS mice. Vamifeport significantly reduced inflammatory markers in HbSS mice: total leukocyte, neutrophil, and lymphocyte counts (A); plasma levels of the chemokine RANTES (B); expression of the neutrophil chemoattractant Cxcl1, as determined by quantitative polymerase chain reaction in liver (C); sVCAM-1 (D); soluble P-selectin (E); and plasma XO activity (F). Results are presented as individual values with mean ± standard deviation (n = 6-8 mice per group). Analysis was performed by using one-way analysis of variance with Dunnett’s multiple comparison of all groups vs the HbSS vehicle group: *P < .05, **P < .01, ***P < .001. BID, twice daily.
Endothelial activation and dysfunction in SCD are associated with increased levels of circulating adhesion molecules.37,38 Consistent with reduced systemic inflammation, vamifeport significantly lowered plasma soluble VCAM-1 (sVCAM-1) and soluble platelet selectin (sP-selectin) levels in HbSS mice (Figure 4D-E). Plasma xanthine oxidase (XO) activity in vehicle-treated HbSS mice was higher than that in HbAA mice (Figure 4F), as previously reported.39 Vamifeport reduced XO activity in HbSS mice, suggesting that iron restriction via ferroportin inhibition diminishes vascular oxidative stress.
Vamifeport prevented organ iron loading
Vamifeport reduced serum iron levels in HbSS mice dosed 3 hours before blood sampling, which is in line with inhibition of ferroportin-mediated iron efflux into blood circulation (Figure 5A). To distinguish the effects of vamifeport on preexisting and newly absorbed iron in organs, mice had access to drinking water containing the stable iron isotope 58Fe during the study. 58Fe concentrations in the liver and 58Fe content in spleen of vamifeport-treated HbSS mice were significantly lower than those of vehicle-treated mice, indicating that vamifeport prevents additional organ iron accumulation (Figure 5B-C). Vamifeport treatment normalized intracellular RBC iron content in HbSS mice to the levels of control HbAA mice (Figure 5D), further emphasizing the potential of vamifeport to minimize the deleterious effects of iron in sickle RBCs.
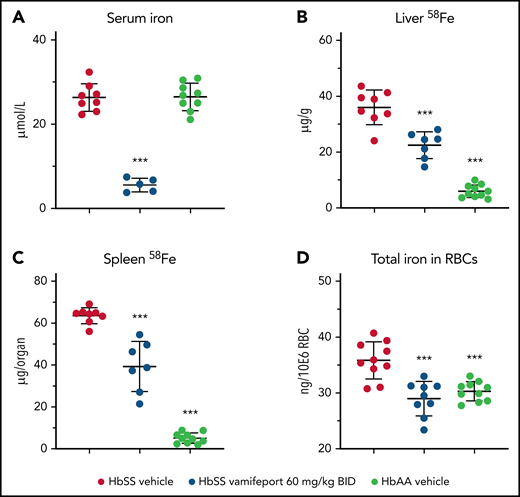
Vamifeport prevented organ iron loading in the Townes model of SCD (HbSS mice). Mice received water supplemented with the stable iron isotope 58Fe to distinguish iron acquired during the treatment period and iron already present in organs. 58Fe concentrations in the liver and spleen were measured by using inductively coupled plasma mass spectroscopy. (A) Vamifeport significantly reduced serum iron levels 3 hours postdose. Vamifeport prevented 58Fe loading of livers (B) and spleens (C) of HbSS mice. (D) Vamifeport corrected total iron content in HbSS RBCs to the levels of HbAA mice. Results are presented as individual values with mean ± standard deviation (n = 7-10 mice per group). Analysis was performed by using one-way analysis of variance with Dunnett’s multiple comparison of all groups vs the HbSS vehicle group: ***P < .001. BID, twice daily.
Vamifeport prevented organ iron loading in the Townes model of SCD (HbSS mice). Mice received water supplemented with the stable iron isotope 58Fe to distinguish iron acquired during the treatment period and iron already present in organs. 58Fe concentrations in the liver and spleen were measured by using inductively coupled plasma mass spectroscopy. (A) Vamifeport significantly reduced serum iron levels 3 hours postdose. Vamifeport prevented 58Fe loading of livers (B) and spleens (C) of HbSS mice. (D) Vamifeport corrected total iron content in HbSS RBCs to the levels of HbAA mice. Results are presented as individual values with mean ± standard deviation (n = 7-10 mice per group). Analysis was performed by using one-way analysis of variance with Dunnett’s multiple comparison of all groups vs the HbSS vehicle group: ***P < .001. BID, twice daily.
Systemic but not dietary iron restriction reduced hemolysis and inflammation
We next asked whether the effects of LID can induce similar pharmacologic effects as ferroportin inhibition using vamifeport. To compare iron restriction by ferroportin inhibition and LID, 6- to 7-week-old HbSS mice were administered either vamifeport or LID for 6 weeks. RBCs of HbSS mice treated with vamifeport, but not LID-fed mice, had hematologic parameters indicating iron-restricted erythropoiesis, with increased percentages of hypochromic and microcytic RBCs (Figure 6A; supplemental Figure 1A, available on the Blood Web site). Intracellular HbS concentration, as measured by CHCM, was significantly reduced in vamifeport-treated HbSS mice but not in LID-fed mice. Plasma indirect bilirubin concentrations were significantly reduced in vamifeport-treated HbSS mice (Figure 6B) but not in LID-fed HbSS mice. Vamifeport, but not LID, reduced markers of systemic and vascular inflammation in HbSS mice (Figure 6C-E). Ferroportin inhibition caused serum iron reduction 3 hours after vamifeport administration, which was not observed in the LID group (Figure 6F), potentially indicating a compensatory mechanism of iron mobilization from stores in LID-fed mice. Indeed, mice given LID had significantly lower total liver iron levels (Figure 6G), which were also evidenced in Perls-stained liver sections (supplemental Figure 1B). These data show that a 6-week LID effectively depleted liver iron in adult Townes mice without affecting erythropoiesis, whereas vamifeport efficiently induced iron-restricted erythropoiesis and liver iron retention. Both ferroportin inhibition and LID lowered liver hepcidin expression (Figure 6H), reflecting feedback responses to the reduction of plasma and liver iron, respectively. Therefore, pharmacologic ferroportin inhibition is required to achieve iron-restricted erythropoiesis (lower hemoglobin), which is not feasible with LID due to chronic suppression of hepcidin synthesis and utilization of iron for production of hemoglobin.

Systemic iron restriction by vamifeport, but not dietary iron restriction, reduced hemolysis and inflammation in HbSS mice. Treatment with vamifeport, but not dietary iron restriction (LID), increased the proportion of circulating hypochromic and microcytic RBCs and reduced CHCM (A) and decreased indirect bilirubin in plasma (B), compared with HbSS mice treated with vehicle. Vamifeport, but not LID, prevented the release of sVCAM-1 into the circulation of HbSS mice (C) and reduced plasma concentrations of RANTES (D). (E) Circulating leukocyte, neutrophil, and lymphocyte numbers were normalized in HbSS mice receiving vamifeport but remained elevated in HbSS mice on LID. (F) Vamifeport, but not LID, decreased serum iron levels. LID, but not vamifeport, efficiently depleted iron from the liver (G), whereas both treatments decreased the expression of liver Hamp (H). Results are presented as individual values with mean ± standard deviation (n = 10-12 mice per group). Analysis was performed by using one-way analysis of variance with Dunnett’s multiple comparison of all groups vs the HbSS vehicle group: *P < .05, **P < .01, ***P < .001. BID, twice daily; QD, once daily.
Systemic iron restriction by vamifeport, but not dietary iron restriction, reduced hemolysis and inflammation in HbSS mice. Treatment with vamifeport, but not dietary iron restriction (LID), increased the proportion of circulating hypochromic and microcytic RBCs and reduced CHCM (A) and decreased indirect bilirubin in plasma (B), compared with HbSS mice treated with vehicle. Vamifeport, but not LID, prevented the release of sVCAM-1 into the circulation of HbSS mice (C) and reduced plasma concentrations of RANTES (D). (E) Circulating leukocyte, neutrophil, and lymphocyte numbers were normalized in HbSS mice receiving vamifeport but remained elevated in HbSS mice on LID. (F) Vamifeport, but not LID, decreased serum iron levels. LID, but not vamifeport, efficiently depleted iron from the liver (G), whereas both treatments decreased the expression of liver Hamp (H). Results are presented as individual values with mean ± standard deviation (n = 10-12 mice per group). Analysis was performed by using one-way analysis of variance with Dunnett’s multiple comparison of all groups vs the HbSS vehicle group: *P < .05, **P < .01, ***P < .001. BID, twice daily; QD, once daily.
Vamifeport reduced vascular adhesion and improved hemodynamics
The deformability of RBCs as a function of oxygen pressure was measured by oxygen gradient ektacytometry (oxygenscan). HbSS mice treated with vamifeport showed similar point of sickling as control HbSS mice (Figure 7A). The point of sickling is defined as the oxygen pressure at which a 5% decrease in the EI is observed during deoxygenation.40 In agreement with these results, image-based analysis of RBC sickling triggered by a progressive decrease in oxygen concentration showed similar percentages of sickle RBCs in blood from HbSS mice treated with either vamifeport or vehicle (supplemental Figure 2). Nevertheless, RBCs from HbSS mice treated with vamifeport exhibited improved deformability under hypoxic conditions, as shown by decreased ΔEI (Figure 7B). ΔEI is the difference between the EIs of RBCs at maximal and minimal oxygenation and reflects the severity of sickling (Figure 7C-D). This improvement was not driven by changes in deformability under full oxygenation, as there was a minor change in the EI at maximal oxygenation (Figure 7D), which was most likely due to the higher percentage of microcytic RBCs in HbSS mice treated with vamifeport. Under hypoxia, however, RBCs of vamifeport-treated HbSS mice had significantly higher EI at minimal oxygenation compared with vehicle-treated mice (Figure 7C), demonstrating a beneficial effect of vamifeport on hypoxia-induced severity of sickling. Scanning electron microscopy analysis revealed visually elongated RBCs with fibrous membranes in vehicle-treated HbSS mice, consistent with a sickle RBC morphology; visually rounder and more biconcave-shaped RBCs were found in HbSS mice treated with vamifeport (Figure 7E-F; supplemental Figure 3).
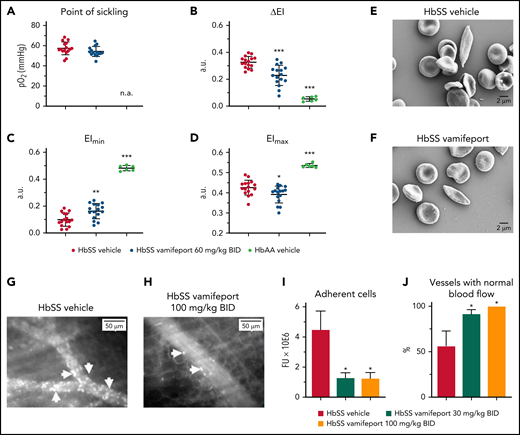
Vamifeport improved deformability of RBCs in hypoxia, reduced the adhesion of blood cells to microvasculature, and prevented inflammation-triggered vaso-occlusion in HbSS mice. (A) No change in the point of sickling of RBCs in HbSS mice; measurement of point of sickling is not applicable (n.a.) to HbAA mice. Reduced severity of sickling shown as a decrease in ΔEI (B) and lowered EI at minimal oxygenation (EImin) (C) with a minor change in the EI at maximal oxygenation (EImax) (D). (E and F) Scanning electron microscopy analysis revealed RBCs with improved membrane morphology in HbSS mice treated with vamifeport compared with vehicle. Representative intravital microscopy images showing marked blood stasis (vaso-occlusion) in capillaries of vehicle-treated Townes mice (arrows show firmly adherent cells) (G), reduced blood cell adhesion in capillaries of vamifeport-treated HbSS mice (H), the number of adherent blood cells (I), and the percentage of vessels with normal blood flow (J). In average, 12 to 35 vessels per mouse in each group were analyzed. Results are presented as mean ± standard deviation (n = 4-7 mice per group). Analysis was performed by using one-way analysis of variance with Dunnett’s multiple comparison of all groups vs the HbSS vehicle group: *P < .05, **P < .01, ***P < .001. a.u., arbitrary units; BID, twice daily; FU, fluorescence units.
Vamifeport improved deformability of RBCs in hypoxia, reduced the adhesion of blood cells to microvasculature, and prevented inflammation-triggered vaso-occlusion in HbSS mice. (A) No change in the point of sickling of RBCs in HbSS mice; measurement of point of sickling is not applicable (n.a.) to HbAA mice. Reduced severity of sickling shown as a decrease in ΔEI (B) and lowered EI at minimal oxygenation (EImin) (C) with a minor change in the EI at maximal oxygenation (EImax) (D). (E and F) Scanning electron microscopy analysis revealed RBCs with improved membrane morphology in HbSS mice treated with vamifeport compared with vehicle. Representative intravital microscopy images showing marked blood stasis (vaso-occlusion) in capillaries of vehicle-treated Townes mice (arrows show firmly adherent cells) (G), reduced blood cell adhesion in capillaries of vamifeport-treated HbSS mice (H), the number of adherent blood cells (I), and the percentage of vessels with normal blood flow (J). In average, 12 to 35 vessels per mouse in each group were analyzed. Results are presented as mean ± standard deviation (n = 4-7 mice per group). Analysis was performed by using one-way analysis of variance with Dunnett’s multiple comparison of all groups vs the HbSS vehicle group: *P < .05, **P < .01, ***P < .001. a.u., arbitrary units; BID, twice daily; FU, fluorescence units.
Intravital microscopy analysis of fluorescently labeled blood cells in the microvasculature of vehicle-treated HbSS mice showed marked vascular adhesion of RBCs and leukocytes, most of which was irreversible during 1 hour of microscopy (Figure 7G). All evaluated venules from HbSS mice treated with vamifeport 100 mg/kg showed normal blood flow (Figure 7H). Cell adhesion in inflamed venules of the vehicle-treated control mice was persistent, resulting in partial or complete blood stasis in >40% of the microvessels (Figure 7I-J; supplemental Video 1 - vamifeport 100 mg/kg and supplemental Video 2 - vehcle). These data suggest that vamifeport improves hemodynamics and prevents vaso-occlusion in the Townes model of SCD.
Discussion
The data presented show a preclinical proof-of-concept for the oral ferroportin inhibitor vamifeport (VIT-2763) in the Townes mouse SCD model. By potently inhibiting ferroportin, vamifeport efficiently induced iron-restricted erythropoiesis, resulting in the development of hypochromic and microcytic RBCs with decreased intracellular HbS concentrations. Importantly, the tested vamifeport doses did not exacerbate anemia, as shown by preserved RBC counts. Vamifeport-treated mice exhibited reduced hemolysis markers, including plasma heme, lactate dehydrogenase, and bilirubin. Notably, vamifeport ameliorated systemic inflammation (reduced circulating leukocytes and RANTES), decreased endothelial activation markers (sVCAM-1 and sP-selectin), reduced oxidative stress, and improved RBC morphology, severity of sickling, and hemodynamics in this SCD model. Based on these findings, we propose that vamifeport mitigates sickling, a downstream effect of which is reduced hemolysis and the amount of cell-free hemoglobin and heme mediators of endothelial damage.
The “iron hypothesis” is based on the concept that lowering RBC HbS concentrations in patients with SCD is associated with decreased HbS aggregation.13,16 Vamifeport lowered RBC HbS concentrations (CHCM) of HbSS mice by ∼5%. This reduction was also evidenced by increased circulating hypochromic cells. Even small decreases in HbS concentrations can have marked inhibitory effects on HbS polymerization9 and increase the delay time of gelation, which is inversely proportional to more than the 30th power of the intracellular HbS concentration.9 Thus, the relatively small effect of vamifeport on HbS concentrations might cause a substantial and clinically relevant delay in HbS polymerization upon deoxygenation, thereby preventing not only hemolysis of circulating RBCs but also vaso-occlusion. In support of this concept, vamifeport improved the deformability of RBCs under hypoxic conditions and RBC membrane morphology, which likely contributes to ameliorated hemodynamics in the Townes model. A model simulating HbS polymerization in conditions of rapidly decreasing oxygen pressure showed that therapeutic reduction of HbS concentrations or increase of fetal hemoglobin has the potential to deliver more oxygen to tissues than drugs that increase the oxygen affinity of hemoglobin.41
Intravascular hemolysis of sickle RBCs is a hallmark of SCD, with multiple downstream effects resulting in anemia, expansion of erythropoiesis, HbS and heme release, increased cell adhesion, inflammation, and vaso-occlusion, with subsequent organ injury.42 Patients with homozygous HbS have severe anemia, which may be further aggravated by excessive iron depletion.8 Therefore, dose-finding studies are necessary to determine the appropriate level of iron restriction to improve hemolysis without exacerbating existing anemia. The use of sensitive iron status biomarkers and RBC indices may help monitor the level of iron restriction more precisely.
Cell-free hemoglobin and heme released by hemolyzed RBCs are considered erythroid damage–associated molecular patterns,36,43 and they activate innate immune pathways through Toll-like receptor 4 and inflammasome, which promote and propagate sterile inflammation and oxidative stress.1,6,35 Heme activates endothelial cells, platelets, and leukocytes to upregulate adhesion molecules, and to release prothrombotic mediators and proinflammatory cytokines, which together orchestrate multicellular adhesion leading to vaso-occlusion, tissue ischemia, and organ injury. Vamifeport-induced iron-restricted erythropoiesis led to microcytic hypochromic RBCs that, after intravascular lysis, released lower amounts of erythroid damage–associated molecular patterns, thus diminishing sterile inflammation and downstream pathologies. For example, vamifeport reduced soluble adhesion molecule levels, indicating reduced vascular activation. Levels of sVCAM-1 in patients with SCD are associated with markers of renal dysfunction, hepatic impairment, and severity of pulmonary hypertension.44 P-selectin mediates leukocyte rolling on vascular endothelium and adhesion of erythrocytes. It also plays a critical role in recruiting leukocytes to inflammatory sites and is a key determinant of microvascular flow in SCD mice.45,46 Blockade of P-selectin with a monoclonal antibody (crizanlizumab) resulted in significantly fewer pain crises in patients with SCD.47,48
Another inflammatory manifestation of SCD is elevated leukocyte counts, which are associated with an increase in pain crises, acute chest syndrome, stroke, and mortality.49 In HbSS mice, leukocyte counts were higher than in HbAA control mice, and vamifeport reduced the number of circulating leukocytes, including neutrophils. Vamifeport also reduced proinflammatory chemokine levels, including the leukocyte attractant RANTES. Therefore, by reducing hemolysis, vamifeport attenuated systemic and vascular inflammation effectors, which are determinants of vaso-occlusion and organ injury.
Vamifeport also reduced XO activity in HbSS mice, in line with findings that XO is a driver of hemolysis in SCD, and XO inhibition by febuxostat was found to improve anemia and decrease hemolysis in mice transplanted with HbSS bone marrow.50,51
Nontransfused patients with SCD do not spontaneously develop systemic iron overload.52 However, microvascular occlusion, RBC sequestration, and splenic infarction can induce iron overload in the spleen, whereas renal iron overload is proportional to the extent of intravascular hemolysis.28,53 HbSS mice accumulate excessive iron in liver, kidneys, and spleen as a result of intravascular and extravascular hemolysis of defective RBCs. Furthermore, anemia causes upregulation of intestinal HIF-2α, resulting in iron overabsorption.19 This was strongly reduced by vamifeport. Cortical kidney iron deposition in SCD results from and correlates with chronic intravascular hemolysis and is involved in renal complications.28,29,54 Coinheritance of α-thalassemia reduces hemolysis in patients with SCD by decreasing HbS concentration and polymerization and protects from proteinuria.55-57 Moreover, nephropathy is associated with premature mortality and limited survival of patients with chronic kidney disease.58 Our results suggest that vamifeport may benefit renal function by preventing reactive iron accumulation in kidney tubules.
In agreement with data on the effects of LID in HbSS mice,20 iron restriction by vamifeport led to reductions in HbS concentration, hemolysis, and vascular activation markers. However, LID significantly increased hematocrit, RBC counts, and MCV in a previous study,20 whereas in most of our experiments, vamifeport reduced hematocrit and MCV without affecting RBC counts. The fundamental difference between these studies is the age of mice at the start of iron-restriction therapy: HbSS mice were LID-fed directly after weaning in the study by Parrow et al,20 whereas vamifeport was administered to mice that had been fed a standard rodent diet for 3 to 4 weeks before study initiation. We therefore propose that the differences in outcome are due to the more preventive design in the study of Parrow et al vs the established disease followed by therapeutic intervention design in our study. In support of this hypothesis, feeding LID to 6- to 7-week-old HbSS mice, which had access to a standard iron diet for 3 to 4 weeks after weaning, failed to induce efficient iron-restricted erythropoiesis and modify ensuing pathologies. Hepcidin is downregulated in iron deficiency and anemia to allow for iron export to plasma via ferroportin. Indeed, both LID and iron restriction by vamifeport downregulated liver Hamp expression in HbSS mice. However, LID resulted in low liver iron and normal plasma iron concentrations, whereas vamifeport reduced plasma iron without significantly changing liver iron concentrations, reminiscent of ferroportin inhibition and iron retention in liver macrophages and hepatocytes.22 Therefore, ferroportin inhibition by vamifeport achieves iron-restricted erythropoiesis, with lower HbS, which is not feasible by LID due to reduced endogenous hepcidin synthesis. The same mechanism may apply to blood-letting therapy, which aims to induce an iron-deficient state in patients with SCD.17,59 Phlebotomy is expected to lower hepcidin levels and therefore promote compensatory iron absorption and utilization for HbS synthesis.
Currently, 4 drugs are approved in the United States for SCD; however, each of these addresses a limited set of SCD complications, and each exhibits frequent undesirable side effects.60 Curative therapies, such as hematopoietic stem cell transplantation or gene therapy, are limited to a small patient population or are not yet available. Therefore, before curative therapies become largely accessible, there is an medical need for medicines that target broader SCD sequelae, such as using combination therapies.42,60 Vamifeport is a novel therapeutic modality that is currently in clinical development and showed good tolerability in healthy volunteers.61 Because vamifeport has a mode of action different from currently approved drugs, it may open further opportunities as a single- or multi-agent therapy in SCD.
Acknowledgments
The authors thank the following colleagues from Vifor Pharma Group for their valuable contributions: Stefan Reim and the chemical development team for providing vamifeport; Maria Wilhelm, Anna-Lena Steck, and Jörg Schmitt, the analytical team for inductively coupled plasma mass spectroscopy and inductively coupled plasma optical emission spectroscopy analysis of tissue iron; and Christian Stegmann for critical review of the manuscript. They thank specially Oswaldo Castro for stimulating discussion and reviewing the manuscript. Special thanks to the animal facility head Marco Franchini and animal caretaker Martin Haenggi. They also thank José María Mateos and Maria-Teresa Colangelo Failla from University of Zurich for scanning electron microscopy sample preparation and imaging and Nadja Stadelmann for scientific illustration support. Editorial assistance with preparing the manuscript for submission was provided by Dawn Batty, Aspire Scientific Ltd., and was funded by Vifor Pharma Group.
Authorship
Contribution: N.N. and V.M. wrote the manuscript with input from N.K. and G.I.; V.M. and F.D. provided guidance in developing the hypothesis and designing experiments; N.N., G.I., R.Z., D.J.S., P.W.B., and R.M.B. proposed and planned experiments, and analyzed and interpreted the results; and A.F., N.K., G.I., R.Z., R.M.B., and C.D. performed experiments and analyzed results.
Conflict-of-interest disclosure: N.N., G.I., A.F., N.K., C.D., V.M., and F.D. are current or past employees of Vifor (International) Ltd and may own equities. N.N., V.M., and F.D. are inventors in patents related to the publication. R.Z. received funding from Vifor (International) Ltd. The remaining authors declare no competing financial interests.
Correspondence: Vania Manolova, Vifor (International) Ltd., Rechenstrasse 37, 9014 St. Gallen, Switzerland; e-mail: vania.manolova@viforpharma.com.
No data were uploaded to public databases. Requests for original data may be submitted to the corresponding author.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal