

Key Points
ISB 1342 exhibits potent killing of primary MM cells and MM cell lines with low sensitivity to daratumumab.
ISB 1342 induced complete MM tumor eradication in 2 in vivo mouse models.

Abstract
Although treatment of multiple myeloma (MM) with daratumumab significantly extends the patient’s lifespan, resistance to therapy is inevitable. ISB 1342 was designed to target MM cells from patients with relapsed/refractory MM (r/r MM) displaying lower sensitivity to daratumumab. ISB 1342 is a bispecific antibody with a high-affinity Fab binding to CD38 on tumor cells on a different epitope than daratumumab and a detuned scFv domain affinity binding to CD3ε on T cells, to mitigate the risk of life-threatening cytokine release syndrome, using the Bispecific Engagement by Antibodies based on the TCR (BEAT) platform. In vitro, ISB 1342 efficiently killed cell lines with different levels of CD38, including those with a lower sensitivity to daratumumab. In a killing assay where multiple modes of action were enabled, ISB 1342 showed higher cytotoxicity toward MM cells compared with daratumumab. This activity was retained when used in sequential or concomitant combinations with daratumumab. The efficacy of ISB 1342 was maintained in daratumumab-treated bone marrow patient samples showing lower sensitivity to daratumumab. ISB 1342 induced complete tumor control in 2 therapeutic mouse models, unlike daratumumab. Finally, in cynomolgus monkeys, ISB 1342 displayed an acceptable toxicology profile. These data suggest that ISB 1342 may be an option in patients with r/r MM refractory to prior anti-CD38 bivalent monoclonal antibody therapies. It is currently being developed in a phase 1 clinical study.
Introduction
Multiple myeloma (MM) is the second most common hematological malignancy worldwide, with 35 500 and 54 600 new cases anticipated in 2025 in the United States and Europe, respectively.1 The emergence of CD38-targeted therapies has significantly prolonged the survival of patients with relapsed/refractory MM (r/r MM) who were treated with ≥2 previous therapies. Daratumumab, a human IgG1 monoclonal antibody targeting CD38, is associated with a median overall survival of 20.1 months in patients refractory to proteasome inhibitors and immunomodulatory drugs.2,3 Mechanistically, daratumumab induces the killing of MM cells via antibody-dependent phagocytosis (ADCP), complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC), and direct apoptosis via FcγRs-mediated crosslinking.4-6 Clinical outcomes have further improved with the approval of daratumumab combinations compared with monotherapy.4,7,8 Despite such progress, most patients continue to relapse because of multiple primary and acquired resistance mechanisms to anti-CD38 therapies.4,9,10 Among those mechanisms, transient downregulation of CD38 expression on the surface of MM cells, which never fully recovers expression, has been observed in patients treated with daratumumab.11 ISB 1342 was therefore designed to be active regardless of CD38 expression and to overcome preexisting resistance to daratumumab’s many mechanisms of action. ISB 1342 was engineered using the BEAT platform (Bispecific Engagement by Antibodies based on the TCR platform)12-14 to target the cluster of differentiation (CD)3-epsilon (CD3ε) and CD38. ISB 1342 aims to treat r/r MM by targeting and depleting CD38+ MM cells via T-cell–redirected killing by crosslinking the CD3ε molecules on T cells and the CD38 molecules on MM cells. This bridging activates T cells in a polyclonal manner, independent of the involvement of a specific antigenic peptide presented on the major histocompatibility class proteins or costimulatory molecules.15,16
Here, we evaluated the ability of ISB 1342 to kill MM cells, which model some resistance mechanisms to daratumumab in patients. We demonstrate that ISB 1342 can successfully induce the killing of MM cell lines in vitro and in vivo, as well as primary MM cells in bone marrow aspirates (BMA) from patients previously exposed to daratumumab, whereas the latter possesses limited activity under these conditions. Studies in cynomolgus monkeys revealed an acceptable toxicology profile and supported the advancement of ISB 1342 into an ongoing phase 1 dose-escalation clinical study in patients with r/r MM.
Material and Methods
Additional detailed methods are presented in the supplemental Materials and methods, available on the Blood website.
Human samples and cell lines
BMA or peripheral blood samples from patients with MM were obtained from University Hospital Geneva, CHU Nantes (MYRACLE cohort17), and Oxford University Hospitals with informed consent under each site's ethical approval. Human peripheral blood mononuclear cells (hPBMCs) and bone marrow mononuclear cells (BMMCs) from healthy donors and patients with MM were isolated using Ficoll gradients. All cell lines were of human origin (from DSMZ or Sigma-Aldrich) and cultured in the media recommended by the supplier.
Redirected lysis (RDL) assay
MM cell lines were labeled with eFluor670 dye (2 μM) or CFSE (1 μM) and cocultured for 48 to 72 hours with hPBMCs at an effector-to-target ratio (E:T) of 10:1 or 5:1 with ISB 1342 or control molecules and additional treatments (soluble CD38, dexamethasone). The MM cell killing was measured as the decrease in the remaining live target cell count after treatment (based on viability dye staining) normalized with the untreated and noneffector cell conditions. The T-cell response was measured as the proportion of live CD8+ T cells expressing CD25, Ki-67, and granzyme B/perforin (supplemental Tables 1-2).
Multiple mode of action killing assay (MMoAK)
hPBMCs were cocultured with MM cell lines previously labeled with eFluor670 (2μM) in medium containing 50% human serum and 100 U/mL hIL-2 at an E:T of 5:1 to enable ADCC, ADCP, CDC, and T-cell–mediated cytotoxicity. Cocultures were then incubated with ISB 1342, daratumumab (Darzalex, Janssen Biotech Inc), or control molecules. The MM cell killing was measured as the decrease in the remaining live target cell count after treatment (based on viability dye staining) normalized with the untreated and noneffector cell conditions. The T-cell response was measured as the proportion of live CD8+ T cells expressing CD25, CD69, and CD107a (supplemental Tables 1-2).

ISB 1342 properties and binding. (A) Schematic 3D representation of ISB 1342, a bispecific antibody based on the BEAT technology with a Fab targeting CD38, an scFv targeting CD3ϵ, and a Fc carrying the LALA (L234A, L235A) mutation. The model was generated using the BioLuminate software (Schrödinger, New York, NY). (B) Mean ± SD of KD determined either on CD38- T cells (n = 12 donors in 3 independent experiments) and MM cell lines, KMS-12-BM (20 measures from n = 14 independent experiments), NCI-H929 (12 measures from n = 5 independent experiments) and MOLP-8 (9 measures from n = 3 independent experiments), or recombinant human proteins CD3ϵδ (n = 5 independent experiments) and CD38 (n = 3 independent experiments). (C) Representative binding of ISB 1342 on CD38– human healthy T cells (mean ± SD of 4 donors) and KMS-12-BM MM cell line (1 representative measurement from 1 experiment). (D) Epitope mapping of daratumumab and ISB 1342 on CD38. Residues in dark red represent the CD38 residues in a 4 Å radius from the daratumumab chain in the crystal structure of 7DHA. Linear peptide mapping by SPR as well as site-directed mutagenesis were used to determine the binding epitope of ISB 1342 on CD38, shown in green on the CD38 chain of crystal structure 7DHA (in beige color). (E) ISB 1342 does not compete with daratumumab and can engage CD38 prebound by daratumumab. Biotinylated human CD38 protein was loaded on a streptavidin SA biosensor. The biosensor with immobilized CD38 was then dipped in a solution of daratumumab in kinetic buffer to reach saturation of the surface. Then, a saturated biosensor was dipped into a premixed solution of daratumumab + ISB 1342 at equimolar concentrations (red curve) or daratumumab only (blue curve). Plots show binding to the sensor tip as a wavelength shift (response, in nm; y-axis) vs time (in sec; x-axis).
ISB 1342 properties and binding. (A) Schematic 3D representation of ISB 1342, a bispecific antibody based on the BEAT technology with a Fab targeting CD38, an scFv targeting CD3ϵ, and a Fc carrying the LALA (L234A, L235A) mutation. The model was generated using the BioLuminate software (Schrödinger, New York, NY). (B) Mean ± SD of KD determined either on CD38- T cells (n = 12 donors in 3 independent experiments) and MM cell lines, KMS-12-BM (20 measures from n = 14 independent experiments), NCI-H929 (12 measures from n = 5 independent experiments) and MOLP-8 (9 measures from n = 3 independent experiments), or recombinant human proteins CD3ϵδ (n = 5 independent experiments) and CD38 (n = 3 independent experiments). (C) Representative binding of ISB 1342 on CD38– human healthy T cells (mean ± SD of 4 donors) and KMS-12-BM MM cell line (1 representative measurement from 1 experiment). (D) Epitope mapping of daratumumab and ISB 1342 on CD38. Residues in dark red represent the CD38 residues in a 4 Å radius from the daratumumab chain in the crystal structure of 7DHA. Linear peptide mapping by SPR as well as site-directed mutagenesis were used to determine the binding epitope of ISB 1342 on CD38, shown in green on the CD38 chain of crystal structure 7DHA (in beige color). (E) ISB 1342 does not compete with daratumumab and can engage CD38 prebound by daratumumab. Biotinylated human CD38 protein was loaded on a streptavidin SA biosensor. The biosensor with immobilized CD38 was then dipped in a solution of daratumumab in kinetic buffer to reach saturation of the surface. Then, a saturated biosensor was dipped into a premixed solution of daratumumab + ISB 1342 at equimolar concentrations (red curve) or daratumumab only (blue curve). Plots show binding to the sensor tip as a wavelength shift (response, in nm; y-axis) vs time (in sec; x-axis).

ISB1342 induces the killing of MM cell lines in vitro via T-cell engagement. (A) Representative confocal image of ISB 1342 (white) at the synapse between T cell (green) and KMS-12-BM MM cell line (blue) acquired with Zeiss LSM 800 inverted confocal microscope, magnification ×40. (B-F) Cytotoxicity of KMS-12-BM MM cell line (B) and T-cell activation (C), proliferation (D), and degranulation (E) after treatment with ISB 1342 or control molecules in the presence (F) or absence (B-E) of soluble CD38 (sCD38) and healthy PBMC (E:T, 5:1) for 72 hours. Data represent the mean ± SD from 3 PBMC donors performed in 2 independent experiments (B-E) or mean ± SD of EC50 from 3 donors that were compared with the condition without sCD38 using a 1-way ANOVA followed by a Dunnett post hoc comparison (F). (G-H) Cytotoxicity of the RPMI8226 MM cell line (G) and cytokine release (H) in the presence of ISB 1342 ± dexamethasone and healthy PBMC (E:T, 10:1) for 48 hours. Data represent mean ± SD of EC50 compared using a paired t test (G) and mean ± SD at the maximum dose of ISB 1342 tested compared using a paired t test (H) from 6 PBMC donors and performed in 2 independent experiments. ∗∗P < .01, ∗∗∗P < .001. ns, not significant. ANOVA, analysis of variance.
ISB1342 induces the killing of MM cell lines in vitro via T-cell engagement. (A) Representative confocal image of ISB 1342 (white) at the synapse between T cell (green) and KMS-12-BM MM cell line (blue) acquired with Zeiss LSM 800 inverted confocal microscope, magnification ×40. (B-F) Cytotoxicity of KMS-12-BM MM cell line (B) and T-cell activation (C), proliferation (D), and degranulation (E) after treatment with ISB 1342 or control molecules in the presence (F) or absence (B-E) of soluble CD38 (sCD38) and healthy PBMC (E:T, 5:1) for 72 hours. Data represent the mean ± SD from 3 PBMC donors performed in 2 independent experiments (B-E) or mean ± SD of EC50 from 3 donors that were compared with the condition without sCD38 using a 1-way ANOVA followed by a Dunnett post hoc comparison (F). (G-H) Cytotoxicity of the RPMI8226 MM cell line (G) and cytokine release (H) in the presence of ISB 1342 ± dexamethasone and healthy PBMC (E:T, 10:1) for 48 hours. Data represent mean ± SD of EC50 compared using a paired t test (G) and mean ± SD at the maximum dose of ISB 1342 tested compared using a paired t test (H) from 6 PBMC donors and performed in 2 independent experiments. ∗∗P < .01, ∗∗∗P < .001. ns, not significant. ANOVA, analysis of variance.

ISB 1342 induces potent killing of cell lines showing reduced sensitivity to daratumumab. (A) Absolute number of specific antibody bound per cell (sABC) indicating the relative CD38 density on MM cell lines. Data represent mean ± SD and were compared using a 1-way ANOVA followed by a Kruskall-Wallis post hoc comparison using KMS-12-BM as a reference. (B-D) Cytotoxicity of MM cell lines in the presence of daratumumab in CDC (B), ADCP (C), and ADCC (D) assays. Data represent mean ± SD of the maximum response from 4 donors in 2 independent experiments (B), from up to 10 donors in 3 independent experiments (C), and from 5 donors in 3 independent experiments (D), which were compared using a 1-way ANOVA followed by a Tukey post hoc comparison. (E) Cytotoxicity of MM cell lines in the presence of ISB 1342 and healthy PBMCs (E:T, 5:1) for 72 hours in a RDL assay. Data represent mean ± SD of EC50 from 6 PBMC donors, compared using a 1-way ANOVA followed by Dunnett post hoc comparison to KMS-12-BM. (F-H) Schematic representation depicting the MMoAK assay, including 4 MoA: T-cell RDL, ADCC, ADCP, and CDC (F). Cytotoxicity of various MM cell lines in the presence of ISB 1342 or daratumumab, healthy PBMC (E:T, 5:1), normal human serum, and rhIL-2 for 48 hours in a MMoAK assay. Data represent mean ± SD of duplicates from 1 representative donor using nonlinear regression analysis (F), the mean ± SD of EC50 from up to 10 PBMC donors per treatment, and the cell line from 6 independent experiments that were compared using a two-way ANOVA and Sidak post hoc test (G-H). ∗P < .05, ∗∗P < .01, ∗∗∗P < .0001, ∗∗∗∗P < .00001.
ISB 1342 induces potent killing of cell lines showing reduced sensitivity to daratumumab. (A) Absolute number of specific antibody bound per cell (sABC) indicating the relative CD38 density on MM cell lines. Data represent mean ± SD and were compared using a 1-way ANOVA followed by a Kruskall-Wallis post hoc comparison using KMS-12-BM as a reference. (B-D) Cytotoxicity of MM cell lines in the presence of daratumumab in CDC (B), ADCP (C), and ADCC (D) assays. Data represent mean ± SD of the maximum response from 4 donors in 2 independent experiments (B), from up to 10 donors in 3 independent experiments (C), and from 5 donors in 3 independent experiments (D), which were compared using a 1-way ANOVA followed by a Tukey post hoc comparison. (E) Cytotoxicity of MM cell lines in the presence of ISB 1342 and healthy PBMCs (E:T, 5:1) for 72 hours in a RDL assay. Data represent mean ± SD of EC50 from 6 PBMC donors, compared using a 1-way ANOVA followed by Dunnett post hoc comparison to KMS-12-BM. (F-H) Schematic representation depicting the MMoAK assay, including 4 MoA: T-cell RDL, ADCC, ADCP, and CDC (F). Cytotoxicity of various MM cell lines in the presence of ISB 1342 or daratumumab, healthy PBMC (E:T, 5:1), normal human serum, and rhIL-2 for 48 hours in a MMoAK assay. Data represent mean ± SD of duplicates from 1 representative donor using nonlinear regression analysis (F), the mean ± SD of EC50 from up to 10 PBMC donors per treatment, and the cell line from 6 independent experiments that were compared using a two-way ANOVA and Sidak post hoc test (G-H). ∗P < .05, ∗∗P < .01, ∗∗∗P < .0001, ∗∗∗∗P < .00001.
Ex vivo assay on samples from patients with MM
Baseline phenotype analysis was performed on 0.2 × 106 to 0.5 × 106 BMMCs and MM cell lines to assess the phenotype of MM cells and T cells (supplemental Table 1). The killing assay was performed on 0.1 × 106 to 0.2 × 106 BMMCS treated with ISB 1342 or daratumumab in medium containing 10% HS and hIL-6 (3 ng/mL) for 17 to 32 hours at 37°C (supplemental Table 2). Tumor cell killing was calculated as the decrease of the remaining live target cell count, defined as CD138+, after treatment, normalized to the untreated condition. The T-cell response was measured as the proportion of live CD8+ T cells expressing CD25, CD69, and CD107a.
In vivo efficacy mouse model
The in vivo study was performed with 6-/7-week-old immunodeficient female NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ) mice (8 mice per group) from Charles River Laboratories and conducted according to the Swiss Animal Protection Law with authorization from the cantonal and federal veterinary authorities. A total of 10 × 106 KMS-12-BM cells were injected subcutaneously, and 10 × 106 hPBMCs were injected intraperitoneally. Treatments were injected IV 9 days later, when tumors reached an average volume of 150 mm3, and then once (ISB 1342) or twice (daratumumab) per week for 3 weeks. Immunoglobulins were injected IV 1 day before each treatment injection. When animals reached maximum tumor size (1000 mm3) before the study end point, they were euthanized, and the last observation carried forward was used. The tumor size was evaluated 3 times per week. For the flow cytometry analysis, tumors were harvested 7 days after the first treatment injection and dissociated with an enzymatic cocktail from a tumor dissociation kit using a GentleMACS dissociator. Cells in suspension were filtered, red blood cells (RBC) were lyzed, and they were stained for human immune cell infiltration and CD38 expression on tumor cells (supplemental Table 1).
Studies in cynomolgus monkeys
Monkey studies were conducted at Shin Nippon Biomedical Laboratories USA Ltd (SNBL), an Association for Assessment and Accreditation of Laboratory Animal Care–accredited facility. Purpose-bred (Cambodian origin), naïve male and female cynomolgus monkeys were used in the non–good laboratory practice study. The study protocol and amendments were approved by the study director and SNBL’s Institutional Animal Care and Use Committee. All procedures were performed in compliance with the SNBL standard operating procedures. cynomolgus monkeys (1 male and 1 female) received a single IV bolus injection at escalating doses of ISB 1342 (1, 100, and 1000 μg/kg at days 1, 29, and 57, respectively). Clinical observations and blood samples were collected for clinical pathology, cytokines, antidrug antibody (ADA), and flow cytometry analysis of leukocyte populations (supplemental Table 1). The serum concentrations of ISB 1342 were measured using an exploratory hybrid IP-LC/MS/MS method at Q2 Solutions (Ithaca, NY).
Results
ISB 1342 engineering and biophysical characterization
The anti-CD3ε scFv portion of ISB 1342 was genetically engineered by fusing the variable heavy chain and light chain domains of a humanized version of the SP34 mouse antibody via a 15-amino acid linker ((Gly4Ser)3). The resulting scFv domain is connected to the hinge region via a short 5-amino acid linker (Gly4Thr). The Fab portion is based on a humanized mouse antihuman CD38 antibody, 9G7, developed by Ichnos and dubbed humanized 9G7 (h9G7). Fc receptors expressed on human peripheral blood cells drive cytotoxic, phagocytic, and inflammatory functions.18,19 To prevent FcγR-mediated binding, which may act as an antibody sink and potentially crosslink FcγR-expressing immune cells with T cells, and to minimize nonspecific T-cell activation (in the absence of target cell engagement), 2 mutations, LALA (L234A/L235A, EU numbering), were introduced into the CH2 domains of ISB 1342 (Figure 1A). These mutations decrease the binding of human IgG1 molecules to human FcγR.20,21 ISB 1342 interacted weakly with all FcγR compared with its Fc-competent counterpart (supplemental Figure 1A-B). ISB 1342 was designed to bind to CD38-expressing tumor cells with high affinity while mitigating the risk of life-threatening cytokine release syndrome (CRS) in the clinic by detuning the affinity to CD3ε to an effective level.22 ISB 1342 binding to human CD38 and CD3εδ recombinant proteins displayed KD of 1.1 ± 0.15 nM and 125 ± 2.8 nM, respectively, when surface plasmon resonance (SPR) results were analyzed using the Langmuir 1:1 model (Figure 1B; supplemental Figure 1C). On MM cell lines expressing different levels of CD38, such as KMS-12-BM (CD38+), NCI-H929 (CD38++), and MOLP-8 (CD38+++), ISB 1342 affinity was higher (KD = 2.5 ± 1.8 nM on KMS-12-BM) than on human CD3+CD38− T cells (KD = 230.4 ± 44.8 nM) (Figure 1B-C). ISB 1342 was designed to target an epitope different from daratumumab, as shown by the nonoverlapping antigen-binding footprints on the 3D structure of CD38 (Figure 1D). The lack of competition was confirmed by the ability of ISB 1342 to bind CD38 despite preincubation of CD38 with daratumumab in a biolayer interferometry assay (Figure 1E).
ISB 1342 induces killing of MM cell lines
We explored the ability of ISB 1342 to specifically kill MM cell lines by engaging T cells in vitro using a RDL assay with hPBMCs as effectors. First, we evaluated the ability of ISB 1342 to mediate synapse formation by confocal microscopy. ISB 1342 was located and enriched at the interface between T cells and KMS-12-BM after at least 4 hours, suggesting the formation of an immunological synapse (Figure 2A; supplemental Figure 2A). Next, we evaluated whether ISB 1342 induced killing of KMS-12-BM cells compared with molecules with 1 or both arms replaced by null arms. ISB 1342 killed with an average half-maximal effective concentration (EC50) of 1.23pM, whereas controls did not induce sufficient killing to calculate an EC50 (Figure 2B). This killing was paired with increased expression of CD25 on CD8+ (Figure 2C) and CD4+ T cells (data not shown), elevated T-cell proliferation (Ki-67 staining), and Granzyme B and Perforin (Figure 1D-E; supplemental Figure 2B). No significant increases for any of these markers were seen with control molecules. Cell staining confirmed efficient binding of ISB 1342 (supplemental Figure 2C) to both cell types, whereas controls bound at similar levels to tumors (CD38-only control) and T cells (CD3-only control). We then evaluated potential T-cell fratricide by ISB 1342. A549 cells expressing CD38 and endothelial growth factor receptor were targeted by a T-cell engager (TCE) with the same CD3 arm as ISB 1342 but targeting endothelial growth factor receptor, which is not expressed on T cells and therefore not expected to induce any T-cell fratricide. In these conditions, both TCE had similar cytotoxic activity and displayed similar counts of CD8+CD38+ T cells, which suggests that ISB 1342 is not inducing T-cell fratricide (supplemental Figure 2D-E). Taken together, these data indicate that ISB 1342 induces coengagement of CD38 on tumor cells and CD3ε on T cells, mediating T-cell activation and killing of tumor cells.
We next tested whether the cytotoxicity of ISB 1342 could be influenced by soluble CD38, which is found in patients with MM at concentrations up to 2.8 ng/mL in serum.23,24 At this concentration, no effect was observed on the cytotoxicity of ISB 1342 (Figure 2F). Patients with MM undergoing treatment with TCE often receive corticosteroids, such as dexamethasone, to treat cytokine-associated toxicities, including CRS.25 Treatment with dexamethasone induced a significant reduction in ISB 1342 cytotoxicity but no change in maximum killing (Figure 2G). In contrast, maximum cytokine release was reduced for TNF-α, IL-6, and IL-2 in the presence of dexamethasone (Figure 2H). Taken together, these data support that ISB 1342–induced cytokine release may be manageable with dexamethasone, whereas maximum killing of tumor cells is sustained.
ISB 1342 induces potent killing of MM cells with low sensitivity to daratumumab
Nihof et al. show that the level of CD38 expression on cells from patients with MM is associated with response to daratumumab therapy.11 To explore the relative impact of CD38 expression on ISB 1342 and daratumumab activities, 4 cell lines with different CD38 expression levels (Figure 3A) were evaluated. KMS-12-BM and NCI-H929 resemble patients with r/r MM with lower CD38 expression, whereas expression on RPMI-8226 and MOLP-8 is high. We noted reduced CDC and ADCP on KMS-12-BM and NCI-H929 compared with MOLP-8 with daratumumab (Figure 3B-C). However, similar ADCC levels were observed with all cell lines (Figure 3D). These observations indicate that KMS-12-BM and NCI-H929 indeed exhibit some resistance features of patient-derived MM cells with reduced sensitivity to daratumumab-mediated killing. With ISB 1342, similar cytotoxicity was observed for all 3 cell lines in a RDL assay (Figure 3E), suggesting that the activity of ISB 1342 does not depend on CD38 expression levels.
To evaluate the combined effect of these observations directly in a single assay, we developed a MMoAK where ADCC, ADCP, CDC, and T-cell–mediated cytotoxicity are enabled (Figure 3F). To achieve this, hPBMCs were cocultured with MM cells in 50% human serum as a source of complement and interleukin-2 (hIL-2) to facilitate natural killer cell functions.26 On KMS-12-BM, NCI-H929, and RPMI8226 (CD38++++), we observed a higher cytotoxicity for ISB 1342 compared with daratumumab (Figure 3F-H), notably with both a lower EC50 in all cell lines tested and a higher maximal killing for NCI-H929 and RPMI8226. ISB 1342 was able to kill the 3 cell lines at similar levels, independently of CD38 expression, and also efficiently activate T cells in this model (Figure 3G; supplemental Figure 3A-B).
Given the use of daratumumab in early lines of therapy, we intended to determine whether cotreatment or pretreatment with this drug could influence the cytotoxicity of ISB 1342 in vitro. In a concomitant MMoAK model (Figure 4A), daratumumab (at a predetermined EC50 = 0.2 nM) did not influence cytotoxicity, maximum killing, or T-cell activation/degranulation of ISB 1342 (Figure 4B-C). In the sequential treatment model (Figure 4D), ISB 1342 potency and T-cell activation/degranulation were also unchanged before and after pretreatment with daratumumab (Figure 4E-F). In both models, the percentage of CD8+CD38+ T cells was higher in the presence of ISB 1342 but not with daratumumab alone, whereas the absolute numbers were reduced with ISB 1342 in the sequential model only (supplemental Figure 3C). These results suggest that at 72 hours, in this model, T-cell viability starts reducing upon activation by ISB 1342, whereas daratumumab is not affecting T cells. Thus, the use of ISB 1342 in daratumumab-pretreated patients after a limited washout period should be possible because residual daratumumab should not interfere with ISB 1342 efficacy.

ISB 1342 in vitro potency is not affected by the concomitant or pretreatment with daratumumab. (A) Schematic representation depicting the MMoAK assay with concomitant treatment with ISB 1342 and daratumumab. (B-C) Cytotoxicity of the NCI-H929 MM cell line (B) and CD8+ T-cell response (C) after treatment with increasing doses of ISB 1342 and a fixed dose of daratumumab or monoclonal antibody control (mAb) in the presence of healthy PBMC (E:T, 5:1), normal human serum, and rhIL-2 for 48 hours. Data represent mean ± SD of EC50 or the maximum response, from 12 donors in 3 independent experiments that were compared using an unpaired t test. (D) Schematic representation depicting the MMoAK assay of pretreatment with daratumumab followed by ISB 1342. (E-F) Cytotoxicity of the NCI-H929 MM cell line (E) and CD8+ T-cell response (F) in the presence of healthy PBMC (E:T, 5:1), normal human serum, and rhIL-2 with increasing doses of ISB 1342 for 48 hours after pretreatment with a fixed dose of daratumumab or monoclonal antibody control for 24 hours. Data represent mean ± SD of EC50 or the maximum response from up to 9 donors in 3 independent experiments that were compared using an unpaired t test; ns, not significant.
ISB 1342 in vitro potency is not affected by the concomitant or pretreatment with daratumumab. (A) Schematic representation depicting the MMoAK assay with concomitant treatment with ISB 1342 and daratumumab. (B-C) Cytotoxicity of the NCI-H929 MM cell line (B) and CD8+ T-cell response (C) after treatment with increasing doses of ISB 1342 and a fixed dose of daratumumab or monoclonal antibody control (mAb) in the presence of healthy PBMC (E:T, 5:1), normal human serum, and rhIL-2 for 48 hours. Data represent mean ± SD of EC50 or the maximum response, from 12 donors in 3 independent experiments that were compared using an unpaired t test. (D) Schematic representation depicting the MMoAK assay of pretreatment with daratumumab followed by ISB 1342. (E-F) Cytotoxicity of the NCI-H929 MM cell line (E) and CD8+ T-cell response (F) in the presence of healthy PBMC (E:T, 5:1), normal human serum, and rhIL-2 with increasing doses of ISB 1342 for 48 hours after pretreatment with a fixed dose of daratumumab or monoclonal antibody control for 24 hours. Data represent mean ± SD of EC50 or the maximum response from up to 9 donors in 3 independent experiments that were compared using an unpaired t test; ns, not significant.
ISB 1342 induces killing of primary MM cells from patients
We evaluated the activity of ISB 1342 and daratumumab in samples from patients not previously treated with daratumumab (dara-naïve), including smoldering MM, newly diagnosed MM, newly diagnosed plasma cell leukemia, and patients at relapse vs patients previously treated with daratumumab (dara-exposed), including heavily treated r/r patients (supplemental Table 3). Both daratumumab and ISB 1342 achieved efficient killing of dara-naïve MM cells, whereas only ISB 1342 was able to achieve efficient killing of dara-exposed MM cells or a single plasma cell leukemia sample (with an EC50 of 77.7 pM) (Figure 5A-D). The viability of MM cells was similar at baseline and after 18 to 24 hours in culture (supplemental Figure 4A-B). Dara-exposed patients had significantly lower CD8+ T cells, natural killer (NK) cells, and monocytes/macrophages to MM cells ratios than dara-naïve patients (Figure 5E; supplemental Figure 4C-E). Thus, the activity of daratumumab correlated with the ratios of NK cells and monocytes/macrophages to MM cells, whereas we observed no correlation between the CD8+ T-cell:MM-cell ratio and ISB 1342 cytotoxic activity (supplemental Figure 4F-H). Counts of CD8+CD38+ T cells were slightly higher in the presence of ISB 1342 compared with daratumumab, and counts of NK cells and monocytes/macrophages were not affected by daratumumab or ISB 1342 treatments (supplemental Figure 5A-C), suggesting an absence of on-target off-tumor killing and T-cell fratricide under these conditions. ISB 1342 was also able to efficiently induce cytotoxicity toward tumor cells from patients with Waldenstrom macroglobulinemia and T-cell acute lymphoblastic leukemia, both expressing low levels of CD38 (supplemental Figure 5D-E). Importantly, all patient samples, irrespective of the group considered, responded to ISB 1342 and showed an increased fraction of activated CD4+ and CD8+ T cells as measured by the increase in CD25+ and CD69+ T cells (Figure 5F-G; supplemental Figure 5F).

ISB 1342 maintains high potency to kill tumor cells from patients previously treated with daratumumab. (A-C) Representative cytotoxicity curves of CD138+ MM cells by ISB 1342 and daratumumab at 18 to 24 hours in samples from patients not previously treated with daratumumab (dara-naïve, patient sample 4) (A), previously treated with daratumumab (dara-exposed, patient sample 15) (B), and dara-naïve plasma cell leukemia (PCL; patient sample 1) (C). Data are mean (A-B) or mean ± SEM of replicates (C) analyzed using nonlinear regression analysis. (D) Maximal cytotoxicity of CD138+ tumor cells with ISB 1342 (10-100 nM) or daratumumab (100 nM) in samples from dara-naïve patients (filled symbols) vs dara-exposed (open symbols). Dots represent individual samples, and data are mean ± SD compared using 1-way ANOVA followed by Dunnett multiple comparison analysis to daratumumab on dara-naïve samples. (E) Radar plot of average values for CD8+ T cells, NK cells, and monocytes/macrophages ratio to CD138+ MM cells in samples from dara-naïve patients (blue) vs dara-exposed ones (pink). (F) Representative CD8+ T-cell activation with ISB 1342 and isotype control measured by flow cytometry with expression of CD69 (blue) and CD25 (pink) in PCL. Data are mean ± SEM of replicates analyzed using nonlinear regression analysis. (G) Maximum T-cell activation (CD25 and CD69) and degranulation (CD107a) on dara-naïve vs dara-exposed patient samples with ISB 1342. Data are mean ± SD compared using unpaired t test.
ISB 1342 maintains high potency to kill tumor cells from patients previously treated with daratumumab. (A-C) Representative cytotoxicity curves of CD138+ MM cells by ISB 1342 and daratumumab at 18 to 24 hours in samples from patients not previously treated with daratumumab (dara-naïve, patient sample 4) (A), previously treated with daratumumab (dara-exposed, patient sample 15) (B), and dara-naïve plasma cell leukemia (PCL; patient sample 1) (C). Data are mean (A-B) or mean ± SEM of replicates (C) analyzed using nonlinear regression analysis. (D) Maximal cytotoxicity of CD138+ tumor cells with ISB 1342 (10-100 nM) or daratumumab (100 nM) in samples from dara-naïve patients (filled symbols) vs dara-exposed (open symbols). Dots represent individual samples, and data are mean ± SD compared using 1-way ANOVA followed by Dunnett multiple comparison analysis to daratumumab on dara-naïve samples. (E) Radar plot of average values for CD8+ T cells, NK cells, and monocytes/macrophages ratio to CD138+ MM cells in samples from dara-naïve patients (blue) vs dara-exposed ones (pink). (F) Representative CD8+ T-cell activation with ISB 1342 and isotype control measured by flow cytometry with expression of CD69 (blue) and CD25 (pink) in PCL. Data are mean ± SEM of replicates analyzed using nonlinear regression analysis. (G) Maximum T-cell activation (CD25 and CD69) and degranulation (CD107a) on dara-naïve vs dara-exposed patient samples with ISB 1342. Data are mean ± SD compared using unpaired t test.
ISB 1342 exhibits antitumor activity in vivo
We evaluated the antitumor activity of ISB 1342 in 2 mouse models. NSG mice were engrafted subcutaneously with KMS-12-BM and injected intraperitoneally with hPBMCs (Figure 6A). In this model, ISB 1342 was able to control tumor growth by day 12, whereas we detected no tumor regression with daratumumab compared with vehicle control (Figure 6B). In addition, an increase in the number of tumor-infiltrating hCD45+ cells and T cells was detected specifically in mice treated with ISB 1342 on day 7 (Figure 6C-D), as expected from TCE mechanism of action.27-30 We also observed a substantial increase in CD25+ and/or CD69+ tumor–infiltrating T cells, reflecting their activated status (Figure 6E). A second model expressing very high levels of CD38 (Daudi) showed similar tumor control to ISB 1342. In this model, daratumumab was able to induce partial control of tumor growth, and no tumor control was detected with the CD3-only control (supplemental Figure 6). These in vivo results show that ISB 1342 tumor cell killing is triggered in vivo independently of CD38 expression levels, unlike daratumumab.

ISB 1342 controlled tumor growth in vivo in the KMS-12-BM xenograft hPBMC-transferred NSG mouse model. (A-B) Experimental design (A) and measurement of tumor growth (B) in the KMS-12-BM subcutaneously xenograft hPBMC-transferred NSG mouse model. In vivo activity was followed for ISB 1342 at 0.5 mg/kg injected IV once per week and daratumumab at 16 mg/kg injected IV twice per week, both for 3 weeks with 8 mice per group. Data are mean (mm3) ± SD determined by caliper measurements. Data were compared for both models using 2-way ANOVA followed by Tukey post hoc comparison. ∗Significant differences between ISB 1342 and vehicle control; # shows differences between daratumumab and ISB 1342. (C-D) Infiltration of hCD45+ cells (defined as live hCD45+mCD45–) (C) and T cells (hTCRαβ+CD14–CD19–CD56–CD45+) (D) in tumors of KMS-12-BM xenografted mice after vehicle, ISB1342 or daratumumab treatments. Data are mean ± SD for 5 mice compared using 1-way ANOVA followed by Dunnett post hoc test to ISB 1342. (E) Representative dot plots showing activation profile (CD25 and CD69 expression) on tumor-infiltrating T-cell activation in vehicle, daratumumab, and ISB 1342-treated mice. (F) CD38 expression on MM cells in tumors (KMS-12-BM model). Data are mean ± SD compared using 1-way ANOVA followed by Tukey post hoc test; ∗P ≤ .05.
ISB 1342 controlled tumor growth in vivo in the KMS-12-BM xenograft hPBMC-transferred NSG mouse model. (A-B) Experimental design (A) and measurement of tumor growth (B) in the KMS-12-BM subcutaneously xenograft hPBMC-transferred NSG mouse model. In vivo activity was followed for ISB 1342 at 0.5 mg/kg injected IV once per week and daratumumab at 16 mg/kg injected IV twice per week, both for 3 weeks with 8 mice per group. Data are mean (mm3) ± SD determined by caliper measurements. Data were compared for both models using 2-way ANOVA followed by Tukey post hoc comparison. ∗Significant differences between ISB 1342 and vehicle control; # shows differences between daratumumab and ISB 1342. (C-D) Infiltration of hCD45+ cells (defined as live hCD45+mCD45–) (C) and T cells (hTCRαβ+CD14–CD19–CD56–CD45+) (D) in tumors of KMS-12-BM xenografted mice after vehicle, ISB1342 or daratumumab treatments. Data are mean ± SD for 5 mice compared using 1-way ANOVA followed by Dunnett post hoc test to ISB 1342. (E) Representative dot plots showing activation profile (CD25 and CD69 expression) on tumor-infiltrating T-cell activation in vehicle, daratumumab, and ISB 1342-treated mice. (F) CD38 expression on MM cells in tumors (KMS-12-BM model). Data are mean ± SD compared using 1-way ANOVA followed by Tukey post hoc test; ∗P ≤ .05.
ISB 1342 shows an adequate profile in pharmacokinetic/pharmacodynamic and safety studies
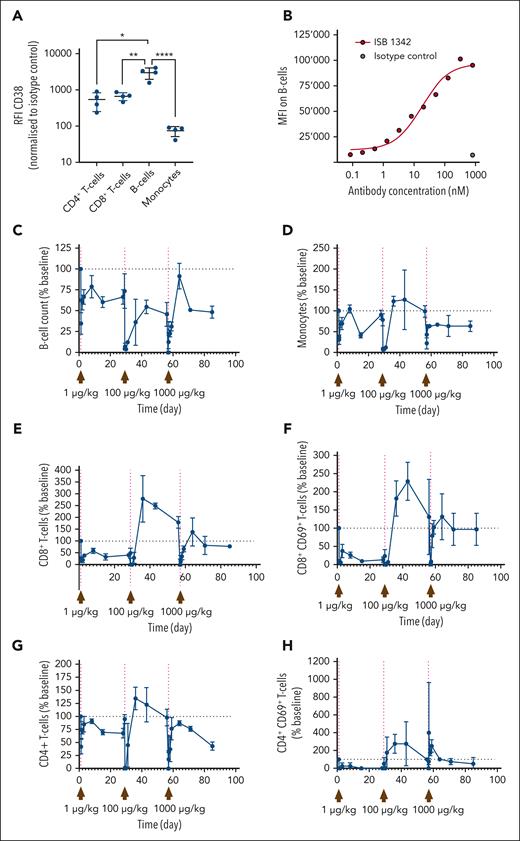
Cancer immunotherapies are often associated with toxicity and tolerability events, generally caused by elevated cytokine release.31 Because CD38 is expressed on human immune cells, in particular on NK, B, and myeloid cells, at similar levels to KMS-12-BM MM cells (supplemental Figure 7A), we therefore examined the ability of ISB 1342 to influence peripheral immune cells in a high-density PBMC assay. Indeed, this assay has been previously reported to increase the sensitivity to T-cell responses and is commonly used for the evaluation of the toxicity of TCE.32,33 In this assay, we did not observe any depletion of peripheral leukocytes in vitro compared with the untreated condition (supplemental Figure 7B). In addition, we found that less ISB 1342 bound to CD38 on RBC compared with daratumumab and observed no sensitization of RBC to hemagglutination compared with positive controls (supplemental Figure 7C-D). We then investigated the pharmacodynamic (PD) changes in peripheral leukocyte populations, cytokine levels, and ISB 1342 PK using cynomolgus monkeys injected IV with consecutive doses of ISB 1342. In cynomolgus monkeys, expression of CD38 on peripheral leukocytes was observed at a significantly higher level on B cells compared with monocytes, CD4+, and CD8+ T cells (Figure 7A); and resulted in detectable levels of ISB 1342 binding to B cells (Figure 7B). One male and 1 female cynomolgus monkeys received ISB 1342 on days 1 (1 μg/kg), 29 (100 μg/kg), and 57 (1000 μg/kg). ISB 1342 induced an initial reduction in B-cell numbers after each dose compared with baseline counts, which rebounded over time but not to baseline levels (Figure 7C). A similar profile was observed for monocytes; however, levels returned to baseline after the first 2 doses (Figure 7D). Such a transient reduction in peripheral populations could reflect the redistribution of these cells rather than their depletion. The number of CD4+ and particularly of CD8+ T cells, including activated CD69+ T cells, substantially increased above the baseline in the circulation of animals administered with 100 μg/kg, indicating T-cell activation and expansion in the periphery (Figure 7E-H). A substantial dose-dependent elevation in serum cytokines, such as IFN-γ was also observed (Figure 7I). ISB 1342 serum concentration profiles followed a biphasic disposition with a short distribution phase followed by a longer terminal elimination phase. The terminal elimination half-life, not confounded by ADA, was ∼4.75 days (Figure 7J). The reduced half-life of ISB 1342 observed at 1000 μg/kg may reflect ADA appearance at this dose (Table 1). These observations were confirmed with the single dose study at 100 μg/kg (supplemental Figure 8; supplemental Table 4). Overall, here, the dose-limiting toxicity was considered to be the CRS, and the maximum tolerated dose was 100 μg/kg using the IV route. ISB 1342 therefore revealed the most common dose-limiting toxicity of TCE and adequate PD and PK profiles to highlight a potential therapeutic window.
Impact of ISB 1342 on circulating leukocytes and systemic soluble factors in cynomolgus monkeys. (A) Expression profile of CD38 on leukocyte populations from cynomolgus monkeys. Dots represent data the relative fluorescence intensity from each measurement and bars represent mean ± SD from 4 animals. Data were compared using a 1-way ANOVA followed by Dunnett post hoc comparison, ∗P ≤ .05. (B) Representative binding of ISB 1342 or isotype control on cynomolgus monkey B cells. (C-H) Cynomolgus monkeys (1 male and 1 female) were injected with 3 consecutive doses of ISB 1342 IV (1, 100 and 1000 μg/kg) at days 1, 29, and 57 respectively. Levels of peripheral B cells (C), monocytes (D), CD8+ T cells (E), activated CD8+ CD69+ T cells (F), CD4+ T cells (G), and activated CD4+ CD69+ T cells (H) were measured using flow cytometry. Data are mean ± SD of 103 counts/μL normalized to baseline counts for 2 animals. (I-J) Levels of circulating IFN-γ (I) and ISB 1342 (J) were measured using ELISA. Data represent levels per animal and LLOQ is the lower limit of quantification for the assay. ELISA, enzyme-linked immunosorbent assay; LLOQ, lower limit of quantitation.
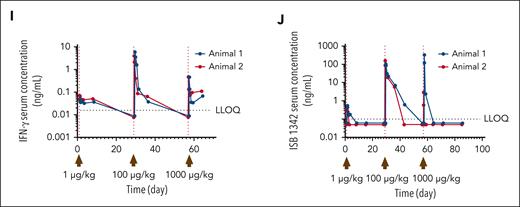
Summary PK parameters of ISB 1342 in cynomolgus monkeys: consecutive-dose study
| Dose level (μg/kg) | C0 (ng/mL) | Cmax (ng/mL) | Tmax (h) | AUC0-t (h × ng/mL) | tlast (h) | AUC0-∞ (h × ng/mL) | t1/2 (h) | ADA detected |
|---|---|---|---|---|---|---|---|---|
| 1 | NE | 0.44 | 4.0 | 8.8 | 36 | NE | NE | None |
| 100 | 206 | 131 | 4.0 | 4589 | 252 | 4863 | 58.2∗ | Day 43 |
| 1000 | 462 | 165.95 | 4.0 | 2193 | 16 | 4355∗ | 2.8 | Day 43 |
| Dose level (μg/kg) | C0 (ng/mL) | Cmax (ng/mL) | Tmax (h) | AUC0-t (h × ng/mL) | tlast (h) | AUC0-∞ (h × ng/mL) | t1/2 (h) | ADA detected |
|---|---|---|---|---|---|---|---|---|
| 1 | NE | 0.44 | 4.0 | 8.8 | 36 | NE | NE | None |
| 100 | 206 | 131 | 4.0 | 4589 | 252 | 4863 | 58.2∗ | Day 43 |
| 1000 | 462 | 165.95 | 4.0 | 2193 | 16 | 4355∗ | 2.8 | Day 43 |
AUC0-∞, area under the (serum) concentration-time curve extrapolated out to infinity; AUC0-t, area under the plasma concentration-time curve from time zero to time t; C0, initial concentration; NE, not estimable; t½, terminal elimination half-life; tlast, time of the last measurable (positive) concentration; Tmax, time to reach maximum serum concentration after drug administration.
N = 1.
Discussion
Advancement in the therapy of MM has substantially improved with the introduction of CD38-targeted monoclonal antibodies.34,35 Daratumumab, the first-approved CD38-targeting monoclonal antibody, has shown significant efficacy in MM36 both as a single therapy37 and as a combination.7,38,39 Despite these results, most patients relapse and become refractory to daratumumab. Though efficacious, isatuximab, the second-approved anti-CD38 monoclonal antibody,40 cannot be used as salvage therapy as it targets CD38 with a similar mode of action, and therefore will be unable to overcome the escape mechanisms of daratumumab treatment.10 The use of TCE, such as ISB 1342, could instead be an option for these patients. The data presented here show that ISB 1342 exhibits more potent cytotoxicity than daratumumab when tested on MM cell lines with varying CD38 expression and lower sensitivity to daratumumab, on patient BMA, or in vivo. Importantly, the potency of ISB 1342 was not affected when combined with daratumumab or in the presence of soluble CD38.
The concept of a CD38 × CD3 TCE12,41 is explored in 2 other disclosed programs: 1) AMG424 (Xencor and Amgen)23 and 2) CD38 × CD28 × CD3 trispecific antibody (Sanofi).42 AMG424 showed good killing activity both in vitro and in vivo. This TCE presents a higher affinity to CD3 (15 nM in SPR) than ISB 1342 and a similar affinity to CD38 (7.7 nM). Unlike the observations we describe here for ISB 1342, AMG424 seems to induce significant depletion of peripheral immune cell populations both in vitro and in vivo.23 The sponsor is currently testing this candidate in the context of T-cell acute lymphoblastic leukemia and acute myeloid leukemia (NCT05038644). The trispecific CD38 × CD28 × CD3 may enhance the potency and persistence of T cells by providing costimulatory signals. The reported in vitro/in vivo results warranted a clinical trial (NCT04401020). Another CD3 × CD38 TCE was described recently and is not currently in clinical development.43 The activity of this TCE seems to depend on the expression levels of CD38 but does not induce depletion of peripheral immune cells. Despite these advantages in terms of the absence of on-target off-tumor activity and the lack of dependency on CD38 levels of ISB 1342 compared with published CD3 × CD38 TCEs, a direct comparison of ISB 1342 to these based on published data is not straightforward without a side-by-side investigation in vitro and in vivo.
ISB 1342 was designed with the advantage that it targets a different epitope from daratumumab to avoid long washout periods. Indeed, a minimal washout period of 3 to 6 months is usually necessary with other anti-CD38 therapies targeting overlapping epitopes or with daratumumab retreatments due to the decrease in CD38 expression and potential competition.11 This delay in treating patients can be problematic and favors the occurrence of resistant clones.44 The key resistance mechanisms described for daratumumab include: downregulation of CD38,4,11 increased expression of complement inhibitors (CD46, CD55, and CD59) limiting CDC,11 and upregulation of CD47, which interferes with phagocytosis.10 Here, we modeled lower sensitivity to daratumumab using cell lines displaying some of these features and patient samples post-daratumumab therapy. In agreement with the literature, the activity of daratumumab was also influenced by the ratio of effector to MM cells in patient samples.45 However, the data presented here show that the mode of action of ISB 1342 is mediated by T cells, making it insensitive to the features of daratumumab resistance such as upregulation of complement inhibitor proteins or CD47. We also show that ISB 1342 can efficiently kill MM cells regardless of CD38 expression, T cell-to-MM cell ratio and recent treatment with daratumumab, providing a rationale for using ISB 1342 in patients relapsing after daratumumab treatment.
Patients with MM showing a T-cell exhaustion profile are more likely to develop progressive disease compared with those with less exhaustion.46 Indeed, patients undergoing autologous stem cell transplantation plus lenalidomide as maintenance therapy exhibit signs of T-cell exhaustion before relapsing.47 Similar preclinical findings were observed in the context of TCEs. In fact, when blinatumomab was continuously administered for 28 days, T cells developed an exhausted phenotype and could not kill target cells, whereas with intermittent dosing, T cells retained their memory TCF1+ phenotype and could control tumor growth in the presence of blinatumomab.48 The quality of T cells defines the activity of TCEs. Hence, some bispecific antibodies are displaying potent cytotoxicity on primary MM, mostly in the presence of healthy T cells,49 but showing reduced activity when exhaustion is detected.50 Several preclinical studies have also shown that a combination with an anti-PD-1 antibody can enhance tumor control by a TCE.51,52 More studies are required to understand how treatment with ISB 1342 will influence T-cell phenotype in the long term, but recent studies demonstrate that patients may benefit from a sequence of 2 different TCE therapies.53 With the recent approval of teclistamab (BCMA TCE, Janssen Biotech) in patients with r/r MM, these findings are key to supporting the development of ISB 1342 in the clinic, which is likely to be administered to patients previously treated with other TCEs. Despite the development and approval of efficient BCMA-targeted therapies, studies have characterized antigen loss, biallelic deletion on chromosome 16 encompassing the BCMA locus, point mutations, shedding, and ADA as mechanisms of resistance to anti-BMCA therapies.54-58 Therefore, it remains essential to monitor biomarkers indicative of these mechanisms and to develop TCE against other validated antigens, such as CD38, to guarantee a range of therapeutic options for patients depending on the features associated with their relapse.
TCE therapies are associated with systemic cytokine release, which is a product of their mode of action. However, this functional cytokine release can progress into CRS, which usually requires intensive care.59-62 Mitigating CRS while maintaining the potential for a beneficial antitumor response is key for TCE therapies, including ISB 1342.63 However, risk factors such as tumor burden and comorbidities often associated with severe CRS should be carefully considered. Corticosteroid treatment is often used to mitigate CRS in the clinic, and data shown here demonstrate that the use of dexamethasone does not strongly affect ISB 1342 cytotoxicity and led to a significant reduction of CRS-associated cytokines in vitro. Using a priming dose or step-up dosing regimen could also mitigate CRS. Indeed, in the teclistamab trial, 40 patients received 1500 μg/kg after 60 μg/kg and 300 μg/kg step-up doses, and no dose-limiting toxicities were observed.64 Finally, the use of monoclonal antibodies before injection of a TCE to reduce peripheral tumor burden could also mitigate CRS. For instance, such an approach was tested for glofitamab,27,65 an anti-CD20 TCE, which was administered after 1 dose of obinutuzumab (anti-CD20 antibody, Roche). This strategy resulted in a manageable CRS while preserving strong potency.66 Whether such approaches could be used for ISB 1342 remains to be clarified in clinical trials. Although preclinical models can assess cytokine release in response to TCE, the field lacks models that accurately predict the occurrence and intensity of CRS in humans. Thus, more studies are warranted to fully assess CRS and to design better options to efficiently mitigate it, such as JAK, mTOR, and Src/Ick inhibitors currently under investigation.67
In conclusion, ISB 1342 is a potent TCE that may be used immediately after or concomitantly with daratumumab to circumvent escape via CD38 downregulation and other mechanisms described previously. Our study suggests that ISB 1342 could elicit antitumor clinical responses in patients with r/r MM who have previously received daratumumab therapy. Based on this encouraging preclinical data and the differentiation from other CD38-targeting therapeutics used in the clinic, a phase 1 clinical trial of ISB 1342 in patients with r/r MM is ongoing (NCT03309111).68
Acknowledgments
The authors thank Camille Grandclément, Evangelia Martini, Valentina Labanca, Stefania De Angelis, Isabelle Gruber, Jérémy Berret, Elodie Stainnack, Riccardo Turrini, Estelle Gerossier, Alain Rubod, Min Ma, Tania Melly, Debora Lind, Paul Oster, Estelle Etasse, and Antoine Job for their technical assistance with the ex vivo and in vivo models; Viviane Villard for providing ISB 1342; and Sunitha Gn (Glenmark) for her work on the IP-LC/MS/MS method. They thank the past Ichnos Sciences SA members, Christelle Ries-Fecourt, Cian Stutz, Amélie Croset, Mégane Pluess, Romain Ollier, and Darko Skegro for their help with developing ISB 1342; Cyrille Touzeau, Nicoletta Lilli, and Sophie Maϊga for access to the patient samples from the MYRACLE (Myeloma Resistance and Clonal Evolution) cohort at CHU Nantes (France), and Cindy Lanvers for her help in collecting patient samples at University Hospital Geneva (Switzerland). The authors are grateful to Sarah Gooding and Mirian Salazar, all patients who donated samples and the HaemBio Biobank, a Medical Research Council and Oxford Biomedical Research Centre funded Biobank, at the MRC Molecular Haematology Unit, Weatherall Institute of Molecular Medicine, University of Oxford, Headley Way, Oxford, OX3 SDS, for the provision of clinical samples. The authors also thank Jairo A. Matthews and Steven M. Kornblau at the University of Texas MD Anderson Cancer Center, Department of Leukemia, Leukemia Sample Bank for access to samples from patients with T-ALL. The authors also acknowledge the Cytocell-Flow Cytometry and FACS core facility (SFR Bonamy, BioCore, Inserm UMS 016, CNRS UAR 3556, Nantes, France) for its technical expertise and help, member of the Scientific Interest Group (GIS) Biogenouest and the Labex IGO program supported by the French National Research Agency (n°ANR-11-LABX-0016-01). Parts of the visual abstract and Figure 3 were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
Authorship
Contribution: B.P., C.E., P.S., E.N., A.L., T.M., A.D., D.P.F., L.C.-I., J.M., J.B., M.C., and M. Perro contributed to assay design, data acquisition, interpretation, and analysis; Z.K., E.M., and C.K. contributed to acquisition of data, analysis, and interpretation of patient samples experiments; T.M., C.P.-D., P.M., C.M.E., J.R.E., A.M., and G.B. contributed to the acquisition of patient samples and discussion of data interpretation; G.S.G., V.U., and V.M. supervised, analyzed, and interpreted the cynomolgus monkey studies; B.P., M.L.M., A.D., M. Pihlgren, M.-A.D., A.S., S.B., E.Z., M.C., and M. Perro contributed to project and resources administration, resources, and supervision; B.P., P.S., C.E., A.D., S.B., A.S., M. Pihlgren, M.-A.D., and E.Z. reviewed and edited the manuscript; and B.P., C.E., M. Perro, and M.C. wrote, reviewed, and edited the manuscript.
Conflict-of-interest disclosure: B.P., C.E., P.S., E.N., A.L., T.M., D.P.F., A.D., L.C.-I., J.M., M. Pihlgren, M.-A.D., G.S.G., V.M., A.S., E.Z., M. Perro, and M.C. are employees of Ichnos Sciences. M. Pihlgren has shares in AC Immune SA. The remaining authors declare no competing financial interests.
Correspondence: Mario Perro, Ichnos Sciences SA, Biopôle Lausanne-Epalinges, Epalinges 1066, Switzerland; e-mail: mario.perro@ichnossciences.com.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal