

Abstract
Reciprocal chromosomal translocations, which are mediated by errors in immunoglobulin heavy chain (IgH) switch recombination or somatic hypermutation as plasma cells are generated in germinal centers, are present in most multiple myeloma (MM) tumors. These translocations dysregulate an oncogene that is repositioned in proximity to a strong IgH enhancer. There is a promiscuous array of nonrandom chromosomal partners (and oncogenes), with the 3 most frequent partners (11q13 [cyclin D1]; 4p16 [FGFR3 and MMSET]; 16q23 [c-maf]) involved in nearly half of MM tumors. It is now shown that a novel t(6;14)(p21;q32) translocation is present in 1 of 30 MM cell lines and that this cell line uniquely overexpresses cyclin D3. The cloned breakpoint juxtaposes gamma 4 switch sequences with 6p21 sequences that are located about 65 kb centromeric to the cyclin D3 gene. By metaphase chromosome analysis, the t(6;14) (p21;q32) translocation was identified in 6 of 150 (4%) primary MM tumors. Overexpression of cyclin D3 messenger RNA (mRNA) was identified by microarray RNA expression analysis in 3 of 53 additional primary MM tumors, each of which was found to have a t(6;14) translocation breakpoint by interphase fluorescence in situ hybridization analysis. One tumor has a t(6;22)(p21;q11) translocation, so that cyclin D3 is bracketed by the IgL and IgH breakpoints. These results provide the first clear evidence for primary dysregulation of cyclin D3 during tumorigenesis. It is suggested that the initial oncogenic event for most MM tumors is a primary immunoglobulin translocation that dysregulates cyclin D1, cyclin D3, and other oncogenes to provide a proliferative stimulus to postgerminal center plasma cells.
Introduction
Dysregulation of oncogenes by translocation near a strong enhancer in an immunoglobulin heavy chain (IgH) (14q32) or IgL (κ, 2p11 or λ, 22q11) locus represents a critical event in the pathogenesis of B-lymphocyte tumors.1,2 Primary translocations usually are mediated by errors in 1 of 3 B cell- and stage-specific DNA modification mechanisms that generate double-strand breaks in genomic DNA: VDJ recombination, IgH switch recombination, and somatic hypermutation.1,3,4 Multiple myeloma (MM) is a tumor of postgerminal center, long-lived plasma cells that have been subjected to each of these 3 DNA modification processes. We and others5-10 have determined that IgH translocations occur in most MM tumors and that they involve a promiscuous array of nonrandom chromosomal partners and oncogenes. The 3 most frequent partner loci, each of which is involved in approximately 10% to 20% of MM tumors, include 4p16.3 (FGFR3 and MMSET), 11q13 (cyclin D1), and 16q23 (c-maf). To gain a more comprehensive insight regarding what kinds of oncogenes are dysregulated by primary immunoglobulin translocations in MM, we have continued to search for additional recurrent translocation partners.
Three D-type cyclin genes encode proteins, each of which can interact with the cdk4 or the cdk6 cyclin D-dependent kinases that phosphorylate Rb, thus regulating a process that promotes the G1/S cell cycle transition.11 Despite differences in expression patterns and in some functional properties, it appears that cyclins D1, D2, and D3 are virtually interchangeable for regulating Rb phosphorylation.12-14 It is well established that dysregulation of cyclin D1 provides a primary oncogenic event in animal models and in various human tumors, including MM.15-23 By contrast, despite overexpression or amplification of cyclins D2 or D3 in some human tumors, there is a paucity of compelling evidence that dysregulation of cyclins D2 or D3 are primary oncogenic events.24-31
We report here the cytogenetic and molecular characterization of a novel partner locus at 6p21 that is translocated to the IgH or Igλ locus in approximately 4% of MM tumors, and identify cyclin D3 as the apparent oncogene that is dysregulated as a consequence of these translocations.
Materials and methods
Probes
The CH and CλBAC, the VH cosmid probe, the 5′ and 3′ switch gamma (Sγ) probes, and the cyclin D3 cosmid (ccnd3cy13) probe have been described elsewhere.8,32,33 The cyclin D3 BAC (RPC11-209e18) was obtained from Research Genetics (Huntsville, AL). For Northern blots, we used cyclin D1 and cyclin D3 cDNA probes.19 34
Cell lines
The MM cell lines used to determine the expression of cyclin D messenger RNA (mRNA) and protein included the following, as described previously6,8 35: RPMI-8226, ANBL6, ARK, Delta-47, EJM, Flam-76, FR4, H1112, H929, JIM-3, JJN-3, Karpas-620, KMM-1, KMS-12, KMS-18, L363, LP-1, MM.1, MM-S1, OCI-MY5, OPM-2, SKMM-1, SKMM-2, U266, UTMC-2, XG-1, XG-2, XG-5, XG-6, and XG-7.
Fluorescence in situ hybridization assays
Three color fluorescence in situ hybridization (FISH) assays of metaphase chromosomes and interphase chromosomes and the 3-color cIg FISH assays have been described in detail previously.8,32 36 For sequential 3-color FISH analyses, after initial hybridization and analysis, slides were rinsed in phosphate-buffered saline, dehydrated in 70%, 90%, and 100% ethanol, stripped in 70% formamide denaturing solution at 72°C for 6 minutes, rehybridized with FISH probes, and then reanalyzed.
Cloning and sequencing of the KMM-1 translocation breakpoint
KMM-1 genomic DNA was digested with HindIII, ligated to BamHI adaptors (New England Biolabs, Beverly, MA), and cloned into λZAP Express vector (Stratagene, LaJolla, CA). A phage clone containing the 6.4-kb translocation breakpoint fragment that hybridizes with 3′Sγ, but not 5′Sγ, probes was isolated. The rescued plasmid was partially sequenced as depicted in Figure2A. The T7 sequence included 317 bp that completely matched sequences upstream of the HindIII site, which is downstream of Sγ4 sequences. The 6236 sequence (accession no. AF364073) started 330 bp 3′ of the Sγ4 region and included the following sequences: Sγ4 3′nt330 > 240/Sγ4 3′nt40 > 232/300 bp of repetitive sequence. The T3 sequence (accession no. AF364072) included 648 bp of repetitive sequence, but 2 oligonucleotides (TATAGGTACCTCCCCAGAAGA and TGCCTGGCTTCTTTCACTTAG) designed from this sequence identified a unique 211-bp fragment from a polymerase chain reaction (PCR) reaction of genomic DNA. This PCR reaction enabled us to identify and isolate the CD3 BAC (see above) that contains cyclin D3 coding sequences and the KMM-1 translocation breakpoint-associated sequences.
Microarray analysis of mRNA in purified normal and malignant plasma cells
Bone marrow plasma cells from 10 healthy donors, 28 patients with newly diagnosed disease, and 25 treated patients with active multiple myeloma were subjected to gene expression analysis using oligonucleotide-based microarrays. Bone marrow cells were aspirated from the iliac crest of healthy donors or patients with active disease during routine clinical work-up. All tests were performed with informed consent, and samples were encrypted to ensure privacy. Mononuclear cells were obtained by Ficoll-Hypaque density gradient centrifugation. Plasma cells were purified by anti-CD138 immunomagnetic bead selection with the AutoMACs system according to the manufacturer's instructions (Miltenyi, Cologne, Germany). Average plasma cell purity, as determined by CD38+/CD45− FACs analysis, was 95.46% (range, 90.58% to 98.85%). Total RNA was isolated, converted to complementary DNA (cDNA), and transcribed into biotinylated complementary RNA (cRNA) and hybridized to the Test2 and HuGenFL oligonucleotide microarrays according to the manufacturer's instructions (Affymetrix, Santa Clara, CA). After hybridization, the hybridization solution was removed, and the GeneChips were installed in a fluidics system for washing and staining with phycoerythrin-streptavidin. The GeneChips were read at a resolution of 6 μm using a Hewlett Packard GeneArray Scanner (Palo Alto, CA). Scanned output files were visually inspected for hybridization artifacts and then analyzed with GeneChip 3.3 software (Affymetrix). All arrays were scaled to an average intensity of 1500 and analyzed independently. The absolute call and the average difference calls of mRNA presence and abundance were performed. The expression value (average difference call) for each gene was determined by calculating the average of differences of intensity (perfect match intensity − mismatch intensity) between probe pairs of the given probe sets. Samples with absent calls or present calls with average difference values less than 54 were given an average difference call of 54 (2.5 × the average raw Q or noise level). The average difference call for the CCND3 gene was compared among all samples.
Other procedures
Results
Cyclin D3 is overexpressed in the KMM-1 MM cell line
Using a cyclin D competitive RT-PCR assay38 and Northern blot analysis, a panel of 30 MM cell lines (M&M) was screened for the expression of cyclin D1, D2, and D3 mRNAs. Cyclin D1 mRNA was present at high levels in 8 cell lines, each of which has a t(11;14) translocation (Flam-76, H1112, KMS-12, MM-S1, SKMM-2, U266, XG-1, and XG-5) but was not detected in most MM cell lines (not shown).19 32 In contrast to the marked variation in cyclin D1 mRNA expression, all 30 MM lines expressed cyclin D3 mRNA (Figure1A and not shown). Although all MM cell lines expressed low and somewhat variable levels of cyclin D3, the KMM-1 MM cell line was exceptional in that it expressed 6 times as much cyclin D3 mRNA as the next highest (H929) of the other 29 cell lines (Figure 1A). Cyclin D3 protein expression paralleled mRNA expression (Figure 1B).

Expression of cyclin D3 in MM cell lines.
(A) Northern blot containing 30 μg (10 μg for sample marked by asterisk) total RNA from each MM cell line was hybridized with a cyclin D3 probe. A portion of the film encompassing the 2.4-kb cyclin D3 mRNA is shown for 15- and 96-hour exposures. Ethidium bromide–stained 2.0-kb ribosomal RNA (rRNA) is shown as a loading control. (B) Western blot containing 100 μg total protein for each of 2 MM cell lines was probed with an anti–cyclin D3 antibody (no. SC-182). A portion of the film containing the 33-kd cyclin D3 band is shown.
Expression of cyclin D3 in MM cell lines.
(A) Northern blot containing 30 μg (10 μg for sample marked by asterisk) total RNA from each MM cell line was hybridized with a cyclin D3 probe. A portion of the film encompassing the 2.4-kb cyclin D3 mRNA is shown for 15- and 96-hour exposures. Ethidium bromide–stained 2.0-kb ribosomal RNA (rRNA) is shown as a loading control. (B) Western blot containing 100 μg total protein for each of 2 MM cell lines was probed with an anti–cyclin D3 antibody (no. SC-182). A portion of the film containing the 33-kd cyclin D3 band is shown.
KMM-1 cell line has a t(6;14)(p21;q32) translocation that juxtaposes the cyclin D3 gene with 3′ IgH enhancer sequences
It was originally reported that the KMM-1 MM cell line has a complex karyotype that includes a14q+ translocation, though the partner chromosome was not identified.39 In addition, there are several reports of t(6;14)(p21;q32) translocations in MM and B-lymphoma tumors.40,41 Because cyclin D3 is located at 6p21, we hypothesized that the KMM-1 cell line would have a t(6;14)(p21;q32) translocation. We used different combinations of VH and CH probes (Figure 2A), a cyclin D3 cosmid probe (Figure 2A), plus chromosome 6 or 14 chromosome painting probes in 3 color-FISH analyses to test this hypothesis. A representative result (Figure 3A) shows a complex karyotype that includes one copy of a normal chromosome 6 plus 5 copies of a rearranged chromosome 6, 2 of which (both der[14]) juxtapose cyclin D3 and 3′ IgH enhancer sequences (CH probe). The overlap of the cyclin D3 and CH signals by interphase FISH (Figure 3B) indicates that the 2 sequences are separated by fewer than 500 kb.42Der(6) t(6;14) with telomeric VH sequences was not detected (not shown). Two additional copies of CH (arrows) are juxtaposed with VH and inserted c-myc sequences, as we have described in detail elsewhere.8

Anatomy of normal and translocated IgH and cyclin D3 loci.
(A) The 1-Mb IgH locus 14q32 includes boxed coding regions, gamma-4 switch region (Sγ4), plus the Eμ and 3′ Εα enhancers (shaded ovals). Vertical arrows indicate HindIII restriction sites. Thick horizontal lines indicate VH cosmid located near the telomere (black dot) and CH BAC probes, plus 5′ and 3′ probes that flank all Sγ regions. For the cyclin D3 gene at 6p21, thick horizontal lines indicate the approximate positions of cosmid and BAC cyclin D3 probes. The anatomy of the KMM-1 der(14) t(6;14)(p21;q32) translocation breakpoint and 6.4-kb HindIII breakpoint clone are also depicted, with horizontal arrows indicating regions that were sequenced (see “Materials and methods”). The diagram is not to scale, and in all cases telomeric sequences are located to the left. (B) Southern blot of the KMM-1 genomic DNA digested withHindIII. The 5′ or 3′ Sγ probes flanking the Sγ regions are indicated at the top of the lanes. The 6.4-kb fragment that uniquely hybridizes with the 3′Sγ probe (*) represents an illegitimate switch recombination fragment that is the translocation breakpoint fragment.
Anatomy of normal and translocated IgH and cyclin D3 loci.
(A) The 1-Mb IgH locus 14q32 includes boxed coding regions, gamma-4 switch region (Sγ4), plus the Eμ and 3′ Εα enhancers (shaded ovals). Vertical arrows indicate HindIII restriction sites. Thick horizontal lines indicate VH cosmid located near the telomere (black dot) and CH BAC probes, plus 5′ and 3′ probes that flank all Sγ regions. For the cyclin D3 gene at 6p21, thick horizontal lines indicate the approximate positions of cosmid and BAC cyclin D3 probes. The anatomy of the KMM-1 der(14) t(6;14)(p21;q32) translocation breakpoint and 6.4-kb HindIII breakpoint clone are also depicted, with horizontal arrows indicating regions that were sequenced (see “Materials and methods”). The diagram is not to scale, and in all cases telomeric sequences are located to the left. (B) Southern blot of the KMM-1 genomic DNA digested withHindIII. The 5′ or 3′ Sγ probes flanking the Sγ regions are indicated at the top of the lanes. The 6.4-kb fragment that uniquely hybridizes with the 3′Sγ probe (*) represents an illegitimate switch recombination fragment that is the translocation breakpoint fragment.

Three-color FISH analysis of the KMM-1 MM line.
(A) CH (green), cyclin D3 cosmid (red), and chromosome 6 painting (purple) probes were hybridized to metaphase chromosomes (blue DAPI counterstain). Two copies of CH and cyclin D3 colocalize (arrowheads) on der(14). Two copies of CH (arrows) colocalize with VH and inserted c-myc (not shown). One copy of cyclin D3 is on normal chromosome 6, and 3 additional copies of cyclin D3 are on rearranged chromosome 6. (B) Interphase nucleus from (A) shows colocalization of 2 sets of CH and cyclin D3 probes (arrowheads).
Three-color FISH analysis of the KMM-1 MM line.
(A) CH (green), cyclin D3 cosmid (red), and chromosome 6 painting (purple) probes were hybridized to metaphase chromosomes (blue DAPI counterstain). Two copies of CH and cyclin D3 colocalize (arrowheads) on der(14). Two copies of CH (arrows) colocalize with VH and inserted c-myc (not shown). One copy of cyclin D3 is on normal chromosome 6, and 3 additional copies of cyclin D3 are on rearranged chromosome 6. (B) Interphase nucleus from (A) shows colocalization of 2 sets of CH and cyclin D3 probes (arrowheads).
KMM-1 t(6;14) translocation breakpoint involves Sγ4 sequences
Most translocations in MM involve IgH switch regions, which usually can be detected as illegitimate switch recombination fragments by our previously described Southern blot assay.6 19 Using this assay, we screened genomic DNA from the KMM-1 cell line and identified a 6.4-kb HindIII fragment that hybridizes uniquely with a 3′ Sγ probe but no other 5′ or 3′ switch probe (Figure 2B). The 6.4-kb illegitimate switch recombination fragment was cloned. Sequence analysis of this clone (M&M) shows that a translocation breakpoint is located near the 3′ end of the Sγ4 region (sequence 6236 in Figure 2A). The IgH sequences include a 170-bp inversion immediately adjacent to the breakpoint plus repetitive sequences (presumably from 6p21) on the other side of the breakpoint. Sequences from the T3 end of the clone also were repetitive, but it was possible to design oligonucleotides to generate a PCR assay that enabled us to isolate a BAC clone (CD3 BAC in Figure 2A). In addition to the KMM-1 breakpoint-associated sequences, the CD3 BAC also contains coding sequences from the cyclin D3 gene. Thus, the breakpoint must be located no more than 200 kb centromeric to the cyclin D3 gene (see Note added in proof).
Detection of t(6;14)(p21;q32) in primary MM tumors
Among 150 karyotypically abnormal MM tumors that had been analyzed by conventional cytogenetic and spectral karyotypic (SKY) procedures, 6 had apparent t(6;14)(p21;q32) translocations.9 10 Metaphase chromosomes from 5 tumors with apparent t(6;14)(p21;q32) translocations were analyzed by 3-color FISH using various combinations of VH, CH, CD3 cosmid, and CD3 BAC probes (Figure 2A), plus chromosome 6 or 14 painting probes. Figure 4A shows an MM tumor (MM2 in Table 1) in which a simple reciprocal t(6;14)(p21;q32) translocation juxtaposes the cyclin D3 gene (CD3 cosmid) with 3′ IgH enhancer sequences (CH probe) on the 2 copies of der(14). The CD3 BAC probe cohybridizes with the CH probe on the 2 der(14) but also with the VH probe on the one copy of der(6), indicating that the breakpoint occurs within the CD3 BAC. Three of the 4 remaining tumors (MM1, MM3, and MM4) were also confirmed to have t(6;14)(p21;q32) translocations for which cyclin D3 is closely juxtaposed to CH probe sequences on both metaphase and interphase chromosomes (summarized in Table 1). The fifth tumor (MM5) is confirmed to have a t(6;14)(p21;q32) translocation, with the cyclin D3 gene and chromosome 14 sequences juxtaposed at the translocation breakpoint. However, there has been a duplication and inversion of sequences detected by the CH probe, but the CH probe is not localized near cyclin D3 at the breakpoint. It is possible that IgH enhancer sequences not detectable by our FISH assays are closely juxtaposed to cyclin D3 at the translocation breakpoint. In any case, at least 4 of 5 tumors with a t(6;14)(p21;q32) translocation detected by conventional cytogenetic or SKY analyses appear to have IgH enhancer sequences closely juxtaposed to the cyclin D3 gene. Together, these results indicate that the t(6;14)(p21;q32) translocation, with close juxtaposition of cyclin D3 and 3′ IgH enhancer sequences, is present in approximately 4% of primary MM tumors.
![Fig. 4. Three-color FISH analyses of IgH, Igλ, and cyclin D3 in MM tumor samples. / (A) MM2 tumor: (Ai) CH (green), cyclin D3 cosmid (red), and chromosome 14 painting (purple) probes were hybridized to metaphase chromosomes (blue DAPI counterstain). Two copies of CH and cyclin D3 probes colocalize on der(14) t(6;14)(p21;q32) (only one der[14] shown). One copy of cyclin D3 is on normal chromosome 6. (Aii) The same metaphase chromosomes as in (Ai) were stripped and rehybridized with CH (green), VH (green), cyclin D3 BAC (red), and chromosome 6 painting (purple) probes. Two copies of CH and cyclin D3 colocalize on der(14), one copy of cyclin D3 is on normal chromosome 6, and one copy of VH and cyclin D3 colocalize on der(6). (Aiii) Interphase nucleus from (Ai) shows colocalization (arrowheads) of CH and cyclin D3 probes. (B) A25 tumor: (Bi) Cλ (green) and chromosome 22 painting (purple) probes were hybridized to metaphase chromosomes (blue DAPI counterstain). One copy of Cλ is on der(6) and no der(22) was seen. (Bii) The same metaphase chromosomes as in (Bi) were stripped and rehybridized with Cλ (green), cyclin D3 cosmid (red), and chromosome 6 painting (purple). One copy of Cλ and cyclin D3 colocalize on der(6) t(6;22)(p21;q11). (Biii) Interphase nucleus from (Bi) shows colocalizing Cλ and cyclin D3 probes (arrowhead). (C) CH (red), cyclin D3 BAC (green), and anti-IgL antibody (blue) were reacted with interphase nuclei from MM tumor P12. One copy of CH and cyclin D3 BAC colocalize (arrowhead) in the nucleus of the MM cell (arrow) that is identified by the anti-IgL antibody blue staining of its cytoplasm.](/view-large/figure/9244790/h81311242004.jpeg)
Three-color FISH analyses of IgH, Igλ, and cyclin D3 in MM tumor samples.
(A) MM2 tumor: (Ai) CH (green), cyclin D3 cosmid (red), and chromosome 14 painting (purple) probes were hybridized to metaphase chromosomes (blue DAPI counterstain). Two copies of CH and cyclin D3 probes colocalize on der(14) t(6;14)(p21;q32) (only one der[14] shown). One copy of cyclin D3 is on normal chromosome 6. (Aii) The same metaphase chromosomes as in (Ai) were stripped and rehybridized with CH (green), VH (green), cyclin D3 BAC (red), and chromosome 6 painting (purple) probes. Two copies of CH and cyclin D3 colocalize on der(14), one copy of cyclin D3 is on normal chromosome 6, and one copy of VH and cyclin D3 colocalize on der(6). (Aiii) Interphase nucleus from (Ai) shows colocalization (arrowheads) of CH and cyclin D3 probes. (B) A25 tumor: (Bi) Cλ (green) and chromosome 22 painting (purple) probes were hybridized to metaphase chromosomes (blue DAPI counterstain). One copy of Cλ is on der(6) and no der(22) was seen. (Bii) The same metaphase chromosomes as in (Bi) were stripped and rehybridized with Cλ (green), cyclin D3 cosmid (red), and chromosome 6 painting (purple). One copy of Cλ and cyclin D3 colocalize on der(6) t(6;22)(p21;q11). (Biii) Interphase nucleus from (Bi) shows colocalizing Cλ and cyclin D3 probes (arrowhead). (C) CH (red), cyclin D3 BAC (green), and anti-IgL antibody (blue) were reacted with interphase nuclei from MM tumor P12. One copy of CH and cyclin D3 BAC colocalize (arrowhead) in the nucleus of the MM cell (arrow) that is identified by the anti-IgL antibody blue staining of its cytoplasm.
Three-color FISH analyses of IgH, Igλ, and cyclin D3 in MM tumor samples.
(A) MM2 tumor: (Ai) CH (green), cyclin D3 cosmid (red), and chromosome 14 painting (purple) probes were hybridized to metaphase chromosomes (blue DAPI counterstain). Two copies of CH and cyclin D3 probes colocalize on der(14) t(6;14)(p21;q32) (only one der[14] shown). One copy of cyclin D3 is on normal chromosome 6. (Aii) The same metaphase chromosomes as in (Ai) were stripped and rehybridized with CH (green), VH (green), cyclin D3 BAC (red), and chromosome 6 painting (purple) probes. Two copies of CH and cyclin D3 colocalize on der(14), one copy of cyclin D3 is on normal chromosome 6, and one copy of VH and cyclin D3 colocalize on der(6). (Aiii) Interphase nucleus from (Ai) shows colocalization (arrowheads) of CH and cyclin D3 probes. (B) A25 tumor: (Bi) Cλ (green) and chromosome 22 painting (purple) probes were hybridized to metaphase chromosomes (blue DAPI counterstain). One copy of Cλ is on der(6) and no der(22) was seen. (Bii) The same metaphase chromosomes as in (Bi) were stripped and rehybridized with Cλ (green), cyclin D3 cosmid (red), and chromosome 6 painting (purple). One copy of Cλ and cyclin D3 colocalize on der(6) t(6;22)(p21;q11). (Biii) Interphase nucleus from (Bi) shows colocalizing Cλ and cyclin D3 probes (arrowhead). (C) CH (red), cyclin D3 BAC (green), and anti-IgL antibody (blue) were reacted with interphase nuclei from MM tumor P12. One copy of CH and cyclin D3 BAC colocalize (arrowhead) in the nucleus of the MM cell (arrow) that is identified by the anti-IgL antibody blue staining of its cytoplasm.
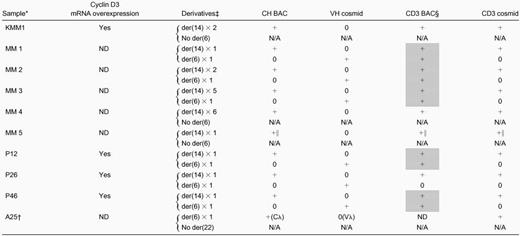
Summary of 6p21 translocations in multiple myeloma tumors
 |
|
ND, not determined; N/A, not applicable.
KMM-1 MM cell line and intramedullary MM primary tumor samples; FISH analyses on interphase nuclei only for P26 and P46, but metaphase chromosomes and interphase nuclei for all other samples.
Variant t(6;22)(p21;q11) translocation.
Type/number of t(6;14)(p21;q32) or t(6;22)(p21;q11) derivatives, with the presence (+) or absence (0) of hybridization signals noted in next 4 columns.
CD3 BAC has cyclin D3 and chromosome 6 KMM1 breakpoint sequences; shaded boxes indicate tumors for which this probe identifies both the der(6) and der(14) derivatives.
Cyclin D3 cosmid and BAC probes are at t(6;14) breakpoint, but CH probe is separated from breakpoint (see “Detection of t(6;14)(p21;q32) in primary MM tumors”).
MM tumor with a t(6;22)(p21;q11) translocation juxtaposing Cλ to cyclin D3
A panel of 30 karyotypically abnormal MM tumors, from which most tumors with an obvious t(11;14) translocation were excluded, was screened by 3-color FISH analyses of metaphase chromosomes for immunoglobulin translocations. No t(6;14)(p21;q32) translocations were identified, but one tumor (A25) with a t(6;22)(p21;q11) translocation was identified (Figure 4B and Table 1). There was close juxtaposition of the Cλ probe, which contains 3′ Cλ enhancer sequences, with CD3 cosmid sequences on the single copy of der(6), but der(22) was not detected. The Cλ and CD3 cosmid sequences consistently overlap on interphase nuclei, indicating that these sequences are separated by fewer than 500 kb. Similar to other oncogenes dysregulated by immunoglobulin translocations, the cyclin D3 gene is bracketed by IgH translocations with centromeric breakpoints and IgL translocations with telomeric breakpoints.1 43
Three of 53 primary MM tumors overexpress cyclin D3 and have t(6;14)(p21;q32) translocations
For each of the primary tumor samples described, we did not have appropriate samples to assess the expression of cyclin D3 mRNA or protein. Recently, however, it became possible to screen purified intramedullary MM tumor cells, which were obtained from 28 untreated and 25 treated patients, for expression of cyclin D3 mRNA by microarray analysis. As seen in Figure 5, MM tumor cells from 3 of 53 patients (P12, P26, P46) showed distinctively high levels of cyclin D3 mRNA expression. Metaphase chromosomes could be obtained only for tumor P12. However, FISH analyses of metaphase chromosomes from P12 and interphase nuclei (Figure 4C) from all 3 tumors demonstrated that the CD3 cosmid probe is closely juxtaposed to the CH probe in each case (summarized in Table1). This confirms the close association of the t(6;14)(p21;q32) translocation with specific overexpression of cyclin D3.

GeneChip HuGeneFL analysis of cyclin D3 expression in MM tumors.
This is a graph of the normalized average difference of fluorescence intensities of cyclin D3 mRNA (vertical axis) in 53 purified MM tumor samples, with each sample represented by a diamond on the horizontal axis. The fluorescence intensity was at background levels for plasma cells from 10 normal donors (not shown).
GeneChip HuGeneFL analysis of cyclin D3 expression in MM tumors.
This is a graph of the normalized average difference of fluorescence intensities of cyclin D3 mRNA (vertical axis) in 53 purified MM tumor samples, with each sample represented by a diamond on the horizontal axis. The fluorescence intensity was at background levels for plasma cells from 10 normal donors (not shown).
Summary of 6p21 translocations with an immunoglobulin locus in MM tumors
Table 1 includes results obtained for the KMM-1 cell line and 9 primary tumors. Eight tumors have a t(6;14)(p21;q32) translocation, with cyclin D3 closely juxtaposed to a CH probe that contains 3′ IgH enhancer sequences in 7 of the tumors. In all informative cases, there is overexpression of cyclin D3. The cell line and 2 tumors have no der(6), whereas the other 6 tumors have one copy of der(6). In contrast, 5 tumors have one copy of der(14), whereas 2 tumors have 2 copies of der(14) and the remaining 2 tumors have 5 or 6 copies of der(14). It is also notable that the KMM-1 cell line and 5 of 6 informative tumors have a t(6;14) translocation breakpoint that occurs within the sequences present in the CD3 BAC clone, so that most breakpoints appear to be clustered in a region that is fewer than 200 kb centromeric to cyclin D3. The single tumor with a t(6;22)(p21;q11) translocation has no der(22), but cyclin D3 is closely juxtaposed to a Cλ probe that contains 3′ Cλ enhancer sequences.
Discussion
Cyclins D1, D2, and D3 are widely expressed in overlapping and apparently redundant patterns, with a degree of tissue specificity for each.11,12,26,29 For example, cyclin D1 is expressed in most proliferating tissues but is not expressed in normal proliferating lymphoid cells that instead express cyclin D2 or cyclin D3.14,31,38,44-47 Cyclin D genes generally are not expressed in quiescent (G0) cells11,34,48-50. In response to growth factors, they are transcriptionally up-regulated and then are expressed in the G1 and sometimes S phases of the cell cycle. Inhibition of cyclin D1 function by antisense sequences or injection of antibodies often results in a complete block of cell proliferation, though this result is not seen in all cases (eg, cells that do not express Rb). In avian DT40 B-lymphoma cells that express cyclins D1 and D2, targeted deletion of the endogenous cyclin D1 gene results in continued proliferation but with a prolonged G1 phase that is rendered normal by the overexpression of transfected cyclin D1, cyclin D2, or cyclin D3 genes.13
Each of the 3 cyclin D genes positively interacts with either the cdk4 or the cdk6 cyclin D–dependent kinases that phosphorylate Rb, resulting in a cascade of events that promote the G1/S cell cycle transition.11 These cyclin D-cdk complexes also bind p27 (Kip1), which does not affect the function of the cyclin D complex, but the sequestration of p27 decreases its ability to negatively regulate cyclin E-cdk2 complexes that are also involved in promoting the G1/S transition.11,51 Notably, the levels of sequestered p27 seem to be higher in lymphomas that overexpress cyclin D3 than in lymphomas that overexpress cyclin D1, suggesting a possible functional difference between cyclin D1 and cyclin D3.52 Cyclin D1 has several cdk-independent activities, including an ability to interact with and partially activate estrogen receptors in the absence of estrogen ligand or more fully activate estrogen receptor–estrogen ligand complexes.29 53 It is unclear whether cyclin D2 or cyclin D3 have cdk-independent activities.
Cyclin D1 is oncogenic in model systems. Overexpression of transfected cyclin D1 in the Rat6 embryo fibroblast cell line has no effect on anchorage dependence, but it causes a decreased duration of G1 and induction of tumors in nude mice after a long latency.18Transfection of rat embryonic fibroblasts with cyclin D1 and H-ras abrogates anchorage dependence and enables them to form fibrosarcomas in nude mice.15,17 Transgenic mice with B-lymphocyte–specific expression of cyclin D1 have apparently normal B cells that do not form tumors, but B-lymphoma tumor formation is enhanced in c-myc, N-myc, or L-myctransgenic mice that also express the cyclin D1 transgene.16,23 An MMTV–cyclin D1 transgene causes abnormal mammary cell proliferation, including development of mammary adenocarcinomas.54
Dysregulation of cyclin D1 can be a primary oncogenic event in human tumors. The best examples of this include the inversion of chromosome 11 and the t(11;14) translocation. The inversion of chromosome 11—inv(11) (p15;q13)—is associated with a substantial increase of cyclin D1 expression in a small fraction of benign parathyroid adenomas,29 and the t(11;14) translocation—which occurs in most mantle cell lymphoma tumors, in approximately 20% of MM tumors, and sometimes in other kinds of B-cell tumors—results in the ectopic expression of cyclin D1, in contrast to the absence of cyclin D1 expression characteristic of normal B cells and most B-cell tumors lacking this translocation.5,19-21,46,55 Cyclin D1 gene amplification, mostly with, but sometimes without, overexpression of cyclin D1, occurs in a substantial fraction of nonhematopoietic tumors, including breast adenocarcinomas, head and neck squamous cell carcinomas, esophageal cancers, hepatocellular carcinomas, and others.29 In some cases, there is overexpression of cyclin D1 without amplification. For breast cancer, amplification and overexpression of cyclin D1 occurs mainly in estrogen receptor-positive tumors.
Convincing evidence that cyclin D2 or cyclin D3 dysregulation can be a primary oncogenic event in model systems or human tumors has been lacking. There is one report of a t(12;22)(p13;q11) translocation in a B-cell tumor with a cloned breakpoint involving a negative regulatory region of the cyclin D2 promoter; it was suggested that this would cause up-regulation of cyclin D2, but no expression data were provided.30 There are limited examples of lymphoid and nonlymphoid tumors with cyclin D2 or cyclin D3 amplification or cyclin D2 or cyclin D3 overexpression, but the significance of this remains unclear.24-29
However, we have now shown that cyclin D3 is dysregulated by translocation to one of the immunoglobulin loci in MM. The t(6;14)(p21;q32) translocation was identified originally by conventional cytogenetics techniques in tumor cells from 2 patients with MM and one patient with plasma cell leukemia. It was also reported as a recurrent translocation that was present in a small number of non-Hodgkin lymphomas.41 Surprisingly, this translocation was not reported by others who used conventional cytogenetics to analyze many karyotypically abnormal MM tumors.56-59Recently, however, we reported 6 tumors with t(6;14)(p21;q32) translocations among 150 karyotypically abnormal primary MM tumors that were analyzed by conventional cytogenetic and SKY procedures.9 10 Conventional cytogenetics identified this translocation in only 3 of the tumors, whereas SKY identified this translocation in all 6 tumors. In “Results” we describe FISH analyses of the KMM-1 MM cell line, 5 MM tumors that have a t(6;14) (p21;q32) translocation identified by conventional cytogenetics or SKY analyses, and 3 MM tumors that overexpress cyclin D3 (Table 1). In the KMM-1 MM cell line and all 6 tumors from which we were able to obtain metaphase chromosomes, cyclin D3 was located at the translocation breakpoint. In KMM-1, in 5 of these 6 tumors, and in the 2 tumors analyzed only by interphase FISH, cyclin D3 was closely juxtaposed to a CH probe that included 3′ IgH enhancer sequences. As found for other oncogenes dysregulated by immunoglobulin translocations, we show that the putative oncogene, cyclin D3, is bracketed by centromeric IgH and telomeric IgL translocation breakpoints. Moreover, cyclin D3 is overexpressed in the cell line (KMM-1) and all 3 informative tumors that have a t(6;14)(p21;q23) translocation, even though all MM cell lines express low levels of cyclin D3 (Figures 1, 5). Our results indicate that immunoglobulin translocations that dysregulate cyclin D3 can be efficiently detected by microarray analysis of RNA expression or by FISH analyses, and we suggest the possibility that microarray analysis of RNA expression may, in fact, be the best way to identify tumors with dysregulated expression of cyclin D3.
Primary immunoglobulin translocations in MM appear to be generated by errors in IgH switch recombination or, less often, by errors in somatic hypermutation that occur during the generation of long-lived plasma cells in germinal centers.60 The 3 major primary translocations in MM involve 4p16, 11q13, and 16q23. Similarly, the t(6;14) (p21;q32) translocation appears to be mediated by an error in IgH switch recombination because the cloned translocation breakpoint in KMM-1 involves a switch region. Given our results for cyclin D3 dysregulation in MM, the similar functional properties of the D-type cyclins and the fact that cyclin D1 is a proven oncogene, it seems clear that cyclin D3 must be oncogenic in MM. However, for cyclin D1, cyclin D3, and other oncogenes dysregulated by translocations in MM, it remains to be determined whether the oncogenic effect results from dysregulated expression in G0 plasma cells or from markedly increased levels of expression in cycling plasma cells.
We thank Johanna Hardin for her help designing methods for analysis of the microarray RNA expression data and Giovanni Tonon for helpful suggestions.
Supported by the Howard Temin Award (CA74265) from the National Cancer Institute (P.L.B.).
J.S. and A.G. contributed equally to this work.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
Note added in proof
The KMM-1 t(6;14) translocation breakpoint is located approximately 65 kb from the 5′ end of the cyclin D3 gene (BAC sequence AL513008).

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal