

Key Points
Imetelstat induces apoptosis of MF but not normal stem/progenitor cells.
Imetelstat is capable of selectively depleting MF stem/progenitor cells.

Abstract
Clinical trials of imetelstat therapy have indicated that this telomerase inhibitor might have disease-modifying effects in a subset of patients with myelofibrosis (MF). The mechanism by which imetelstat induces such clinical responses has not been clearly elucidated. Using in vitro hematopoietic progenitor cell (HPC) assays and in vivo hematopoietic stem cell (HSC) assays, we examined the effects of imetelstat on primary normal and MF HSCs/HPCs. Treatment of CD34+ cells with imetelstat reduced the numbers of MF but not cord blood HPCs (colony-forming unit–granulocyte/macrophage, burst-forming unit–erythroid, and colony-forming unit–granulocyte/erythroid/macrophage/megakaryocyte) as well as MF but not normal CD34+ALDH+ cells irrespective of the patient’s mutational status. Moreover, imetelstat treatment resulted in depletion of mutated HPCs from JAK2V617F+ MF patients. Furthermore, treatment of immunodeficient mice that had been previously transplanted with MF splenic CD34+ cells with imetelstat at a dose of 15 mg/kg, 3 times per week for 4 weeks had a limited effect on the degree of chimerism achieved by normal severe combined immunodeficiency repopulating cells but resulted in a significant reduction in the degree of human MF cell chimerism as well as the proportion of mutated donor cells. These effects were sustained for at least 3 months after drug treatment was discontinued. These actions of imetelstat on MF HSCs/HPCs were associated with inhibition of telomerase activity and the induction of apoptosis. Our findings indicate that the effects of imetelstat therapy observed in MF patients are likely attributable to the greater sensitivity of imetelstat against MF as compared with normal HSCs/HPCs as well as the intensity of the imetelstat dose schedule.
Introduction
Primary myelofibrosis (PMF) as well as post essential thrombocythemia (ET) or polycythemia vera (PV) related myelofibrosis (MF) are characterized by profound structural remodeling of the marrow, megakaryocytic hyperplasia and dysplasia, marrow fibrosis, cytopenias, splenomegaly because of extramedullary hematopoiesis, and disabling systemic symptoms. Advanced forms of each form of MF are associated with limited survival. Approximately 90% MF patients harbor mutations in either JAK2 (58%), calreticulin (CALR, 25%), or myeloproliferative leukemia virus oncogene (MPL, 7%), which each activate JAK-STAT signaling.1-3 MF originates at the level of the hematopoietic stem cell (HSC).4 Except for allogeneic HSC transplantation, however, currently available therapies including the JAK1/2 inhibitor ruxolitinib do not eliminate MF HSCs.
Telomerase is a ribonuclear protein complex made up of a reverse transcriptase catalytic protein subunit (human telomerase reverse transcriptase [hTERT]), an RNA template (hTR), and additional specialized proteins (eg, dyskerin) that act in concert to extend the length of telomeres.5-7 Telomerase is transiently activated in normal stem and progenitor cells but is inactive in mature somatic cells that make up the vast majority of human tissues. Telomerase has, however, been shown to be activated in most cancer cells, irrespective of tumor type.8,9
Imetelstat is a 13-mer oligonucleotide complimentary to the template region of the telomerase RNA component. It binds with high affinity to the template region of the RNA component of telomerase, resulting in direct, competitive inhibition of telomerase enzymatic activity. Preclinical studies have indicated that imetelstat inhibits telomerase activity (TA) and cell proliferation of several cancer cell lines and human tumors in mouse xenograft models.10-17 Moreover, imetelstat also inhibits proliferation and induces apoptosis of cancer stem cells.18,19 Imetelstat has been evaluated in phase 1 and 1/2 clinical trials in patients with solid tumors (eg, breast and lung cancer)20,21 and hematologic malignancies (eg, chronic lymphoproliferative diseases,22 refractory and relapsed multiple myeloma,23 and myeloproliferative neoplasms [MPN]).24,25 Although in the majority of these studies imetelstat has failed to show meaningful clinical activity, phase 2 studies have demonstrated that imetelstat can elicit hematologic and molecular responses in patients with ET who are intolerant or unresponsive to prior standard therapies.24 Moreover, an additional clinical trial in MF patients revealed that imetelstat therapy could achieve complete clinical remissions in some patients using well-established clinical criteria.25 Treatment with imetelstat led to the reversal of bone marrow (BM) fibrosis and induction of morphologic and molecular remissions in some patients with MF.25 These results suggest that imetelstat has disease-modifying activity. The precise mechanism by which imetelstat induces such responses in MPN patients has been poorly defined to date. In this report, we investigated whether imetelstat selectively targets MF HSCs and hematopoietic progenitor cells (HPCs).
Materials and methods
Drugs
Imetelstat sodium (GRN163L) is a 5′ palmitoylated 13-mer thiophosphoramidate oligonucleotide composed of the sequence 5′‐TAGGGTTAGACAA‐3′. Mismatched oligonucleotide (MM) is a 5′ palmitoylated 13‐mer thiophosphoramidate oligonucleotide composed of the sequence 5′‐TAGGTGTAAGCAA‐3′. Both compounds were provided initially by Geron Corporation (Menlo Park, CA) and subsequently by Janssen Research & Development LLC (Raritan, NJ).
Patient specimens and cell preparation
All patients signed informed consents approved by the Institutional Review Board of the Icahn School of Medicine at Mount Sinai (ISMMS). Single-cell suspensions were prepared according to the method of Barosi et al26 from the surgically removed spleens of 13 patients (supplemental Table 1, patients [Pts] 1-10, 16-18) with advanced forms of MF requiring therapeutic splenectomy. The characteristics of these patients and their clinical outcomes have been previously reported.27 peripheral blood (PB) was collected from 5 patients (supplemental Table 1, Pts 11-15) with PMF or PV/ET-related MF who fulfilled World Health Organization diagnostic criteria.28 The JAK2, CALR, and MPL mutational status1,29-31 of each of these patients is shown in supplemental Table 1. Cord blood (CB) collections were provided by the New York Blood Center. CD34+ cells were selected from mononuclear cells using a CD34+ cell selection kit (StemCell Technologies, Vancouver, BC, Canada). CD34+ cells with a purity of ≥90% as analyzed using a FACSCanto Flow Cytometer (BD, Franklin Lakes, NJ) were used in each experiment.
Treatment of MF and normal CD34+ cells with imetelstat
MF or CB CD34+ cells (2.5 × 104/mL) were incubated in serum free expansion medium (StemCell Technologies) supplemented with 50 ng/mL stem cell factor, 100 ng/mL FLT-3 ligand, 100 ng/mL thrombopoietin, and 50 ng/mL interleukin-3 (Gemini Bio-Products, West Sacramento, CA) in the presence of imetelstat or MM (1.8 μM, 3.75 μM, 7.5 μM) or vehicle alone. Seven days after the treatment, the numbers of cells were enumerated and stained with CD34 and a lineage cocktail monoclonal antibodies (mAbs). Moreover, aldehyde dehydrogenase (ALDH) activity of cells harvested was assessed using an Aldefluor kit (StemCell Technologies) according to the manufacturer’s recommendations, followed by staining with a CD34 mAb. All antibodies were purchased from Becton Dickinson (BD) Biosciences (San Diego, CA). Data were acquired using a FACSCanto II Flow Cytometer (BD). Two days after the treatment with imetelstat or MM (7.5 μM), the percentage of CD34+ cells undergoing apoptosis was determined as previously described.32
HPC assays
A fraction of cells harvested from the previous cultures were also analyzed in methylcellulose to which a cytokine cocktail was added according to the manufacturer’s instructions (StemCell Technologies). The numbers of colonies were enumerated after 12 to 14 days of incubation. Individual colony-forming unit–granulocyte/macrophage (CFU-GM) colonies (14-31 colonies per treatment group per patient) were plucked and analyzed for the presence of JAK2V617F using a nested allele-specific polymerase chain reaction (PCR).33 The percentage of JAK2V617F+ CFU-GM was then determined.
Treating NOD/SCID/IL2Rγnull (NSG) mice transplanted with normal or MF splenic CD34+ cells directly with vehicle alone, MM, or imetelstat
NSG mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All experiments were approved by the Animal Care Committee of ISMMS. Initially, in order to identify the dose of imetelstat that was tolerated by NSG mice and that minimally affected the behavior of normal CD34+ cells, CB CD34+ cells from 8 to 10 donors were pooled and were transplanted (5 × 105 per mouse) via the tail vein into 8- to 9-week-old sublethally irradiated (240 cGy) NSG mice. These mice were then injected a week after transplantation intraperitoneally with 5, 15, and 30 mg/kg of imetelstat or MM thrice weekly for 4 to 8 weeks. Two to 3 months after the discontinuation of imetelstat or MM administration, the mice were euthanized and the cells were recovered from the BM of the femurs, tibias, and humeri. The presence of human (h) CD45+, CD14+, CD33+, CD41a+, CD19+, CD3+, and CD34+ cells was determined by mAb staining and flow cytometric analysis.
In order to examine the effects of imetelstat on MF HSCs, MF splenic CD34+ cells (3 × 105 to 5 × 105 per mouse, n = 3) that had previously been shown to achieve significant degrees of human cell chimerism 4 months after their transplantation into NSG mice were used. CD34+ cells from these spleens were transplanted into NSG mice and after a week were treated with imetelstat or MM at the dose of 15 mg/kg for 4 weeks. Three months after the discontinuation of drug treatment, the presence of cells belonging to various human hematopoietic cell lineages in the BMs of recipient mice was quantitated as described previously. In addition, the hCD45+ cells in the BM of the recipient mice were selected using a FACSAria cell sorters (BD). The percentage of JAK2V617F/JAK2total present in the genomic DNA of selected hCD45+ cells from the mice receiving splenic CD34+ cells from a patient with a granulocyte JAK2V617F allele burden of 85.1% was determined using a quantitative real-time (RT)–PCR with an allelic discrimination method.30,31 We considered human engraftment to have occurred in NSG mice if hCD45+ cells were present at ≥0.1% of the nucleated cells in murine BM.
TA assays and telomere length analysis
A quantitative telomerase detection kit (QDT, Allied Biotech, Inc., Benicia, CA) was used to measure TA according to the manufacturer’s instructions which were detailed in the supplemental Methods. For analysis of telomere length, a flow-fluorescence in situ hybridization (Flow-FISH) was performed with a Telomere PNA Kit/FITC for Flow Cytometry (Agilent, Santa Clara, CA) (see supplemental Methods).
Statistical analysis
Results are reported as the mean ± standard deviation. Statistical significance was determined using a 2-tailed Student t test. All P values were 2 sided, and P < .05 was considered significant.
Results
Imetelstat inhibits the proliferation and differentiation of MF but not normal CD34+ cells
Similar numbers of Lin−CD34+ cells (Figure 1A) and assayable HPCs (CFU-GM + burst-forming unit–erythroid [BFU-E] + colony-forming unit–granulocyte/erythroid/macrophage/megakaryocyte [CFU-GEMM]; Figure 1B) were generated when CB CD34+ cells were cultured for 7 days with cytokines alone or cytokines plus increasing doses of either imetelstat or MM (1.8 μM, 3.75 μM, 7.5 μM) (P all > .05). We also assessed the effect of imetelstat on normal CD34+ cells expressing ALDH activity. ALDH activity was used as a surrogate marker for primitive HSCs.34-37 As shown in Figure 1C, similar numbers of CD34+ cells expressing ALDH activity were produced under each of the culture conditions evaluated. By contrast, the treatment of MF CD34+ cells with 7.5 μM imetelstat led to a significant reduction in the numbers of Lin−CD34+ cells, HPCs (CFU-GM + BFU-E + CFU-GEMM) and CD34+ALDH+ cells as compared with MF cells treated with an equal dose of MM (Figure 1D-F). These findings suggest that MF CD34+ cells are more sensitive to the inhibitory actions of imetelstat than normal CD34+ cells.
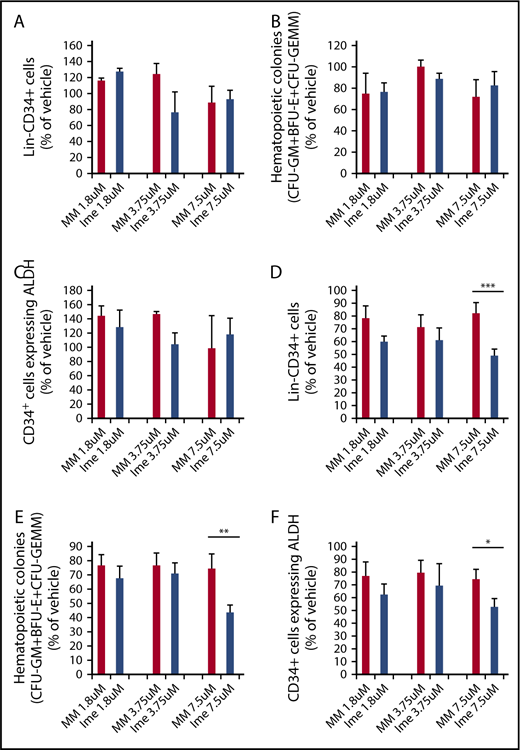
Imetelstat inhibits the proliferation of MF stem and progenitor cells but has limited effects on their normal counterparts. Normal CB (A-C) or MF (D-F) CD34+ cells were treated with cytokines alone or cytokines plus increasing doses of an MM or imetelstat (Ime) for 7 days. Cells generated were phenotypically characterized and were assayed for HPCs. The percentage of the absolute number of Lin−CD34+ cells (A,D), all classes of assayable HPCs (B,E), as well as CD34+ cells with ALDH activity (C,F) generated in the cultures of normal or MF CD34+ cells exposed to cytokines plus MM or cytokines plus Ime relative to that generated in the cultures exposed to cytokines alone are shown. CB: 1.8 μM and 3.75 μM: n = 4; 7.5 μM: n = 6. P all > .05, MM vs Ime at each dose. MF: *P < .05, **P < .01, ***P < .001, MM vs Ime. n = 14 (9 splenic MF and 5 PB MF).
Imetelstat inhibits the proliferation of MF stem and progenitor cells but has limited effects on their normal counterparts. Normal CB (A-C) or MF (D-F) CD34+ cells were treated with cytokines alone or cytokines plus increasing doses of an MM or imetelstat (Ime) for 7 days. Cells generated were phenotypically characterized and were assayed for HPCs. The percentage of the absolute number of Lin−CD34+ cells (A,D), all classes of assayable HPCs (B,E), as well as CD34+ cells with ALDH activity (C,F) generated in the cultures of normal or MF CD34+ cells exposed to cytokines plus MM or cytokines plus Ime relative to that generated in the cultures exposed to cytokines alone are shown. CB: 1.8 μM and 3.75 μM: n = 4; 7.5 μM: n = 6. P all > .05, MM vs Ime at each dose. MF: *P < .05, **P < .01, ***P < .001, MM vs Ime. n = 14 (9 splenic MF and 5 PB MF).
Inhibitory effects of imetelstat on MF CD34+ cells are independent of JAK2V617F or CALR mutational status
Tefferi et al25 suggested that the clinical response to imetelstat therapy occurred exclusively in MF patients who were JAK2V617F+. We, however, demonstrated that the inhibitory effects of imetelstat in vitro were independent of the MF patient’s driver mutational status (supplemental Figure 1).
Effect of imetelstat treatment on malignant MF HPCs
To assess the effect of imetelstat on malignant MF HPCs, individual colonies cloned from cells generated from CD34+ cells from 6 individual JAK2V617F+ MF patients (JAK2V617F allele burden: 25% to 90%) treated with cytokines alone or cytokines plus imetelstat or MM (7.5 μM) were plucked and genotyped. Imetelstat treatment did not affect the percentage of JAK2V617F+ colonies generated when 100% of the colonies contained the mutated form of JAK2 (Table 1). In 3 of 4 patients, however, who had a reservoir of wild-type JAK2 colonies prior to treatment, exposure to imetelstat reduced the percentage of total JAK2V617F+colonies and homozygous JAK2V617F+ colonies (Table 1). Moreover, imetelstat treatment reduced the absolute number of total JAK2V617F+ colonies by 45.8 ± 22.5% (P = .08) and the absolute number of homozygous JAK2V617F+ colonies by 53.6 ± 19.3% (P < .05) (Figure 2). These data suggest that imetelstat treatment is capable of depleting malignant HPCs from a subset of JAK2V617F+ MF patients.
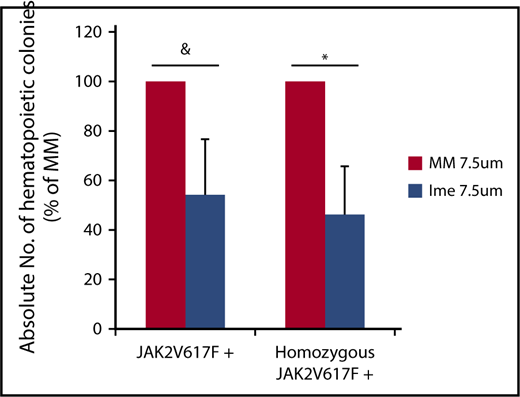
Imetelstat treatment leads to depletion of malignant HPCs. The absolute number of total JAK2V617F+ and homozygous JAK2V617F+ CFU-GM was calculated by multiplying the total number of CFU-GM by the fraction of JAK2V617F+ or homozygous JAK2V617F+ CFU-GM (Table 1) generated in cultures of JAK2V617F+ splenic or PB MF CD34+ cells treated with cytokines alone or cytokines plus MM or Ime (7.5 μM) for 7 days. The percentage of the absolute number of total JAK2V617F+ and homozygous JAK2V617F+ CFU-GM generated in cultures of treated with cytokines plus Ime relative to that generated in cultures treated with cytokines plus MM is shown. n = 4. &P = .08, *P = .03.
Imetelstat treatment leads to depletion of malignant HPCs. The absolute number of total JAK2V617F+ and homozygous JAK2V617F+ CFU-GM was calculated by multiplying the total number of CFU-GM by the fraction of JAK2V617F+ or homozygous JAK2V617F+ CFU-GM (Table 1) generated in cultures of JAK2V617F+ splenic or PB MF CD34+ cells treated with cytokines alone or cytokines plus MM or Ime (7.5 μM) for 7 days. The percentage of the absolute number of total JAK2V617F+ and homozygous JAK2V617F+ CFU-GM generated in cultures of treated with cytokines plus Ime relative to that generated in cultures treated with cytokines plus MM is shown. n = 4. &P = .08, *P = .03.
Effects of treatment with imetelstat on normal NSG repopulating cells (SRCs)
We next examined the effect of imetelstat on normal HSCs by directly treating NSG mice transplanted with CB CD34+ cells with vehicle alone, imetelstat, or MM 1-week posttransplantation. In Figure 3A, representative FACS plots showing hCD45+ and hCD34+ cells generated in the marrow of recipient mice receiving the same CB graft treated with vehicle alone, imetelstat, or MM each at 15 mg/kg are shown. To more accurately assess the effect of imetelstat on normal HSCs, we calculated the absolute number of hCD45+ and hCD34+ cells present in the marrow of the recipient mice by multiplying the total number of marrow cells harvested from 2 tibias, 2 femurs, and 2 humeri by the percentage of hCD45+ and hCD34+ cells detected, respectively. As shown in Figure 3B, 3 months after the completion of the 4 weeks of treatment, the mice transplanted with CB CD34+ cells and treated with MM at each dose tested did not exhibit a decrease in the degree of hCD45+ marrow cell chimerism. However, treatment with 15 mg/kg imetelstat led to a modest reduction in the number of hCD45+ cells (20.9 ± 20.4% vs vehicle alone; 40.4 ± 28.7% vs MM). A slightly greater reduction was associated with the 30 mg/kg dose of imetelstat administration. The numbers of hCD34+ cells were only reduced in mice treated with imetelstat at the highest dose studied (30 mg/kg) (10.8 ± 7.0% vs the equal dose of MM and 38.0 ± 9.7% vs vehicle alone) (Figure 3C). These findings indicate that imetelstat treatment can affect normal hematopoiesis in a dose-dependent fashion. In addition, as shown in supplemental Figure 2, the absolute number of hCD45+CD41a+ megakaryocytes in the mice receiving 30 mg/kg imetelstat was reduced by 50.0% to 78.7% and 18.2% to 42.0% as compared with mice receiving vehicle alone and the same dose of MM in 2 of the 3 normal CD34+ cell samples transplanted, respectively. We next assessed whether prolonged treatment with imetelstat would lead to greater inhibition of normal HSCs. Treatment of the mice transplanted with normal CD34+ cells with 15 mg/kg imetelstat for 8 weeks resulted in a 15.4 ± 6.6% reduction in the number of hCD34+ cells, which was not observed after 4 weeks of treatment (Figure 3E). Additionally, a greater reduction in the degree of hCD45+ cell chimerism and the number of hCD34+ cells was not observed with more prolonged treatment with the 30 mg/kg imetelstat dose (Figure 3D-E). The higher doses of imetelstat, however, led to a 9.0% to 15.6% reduction in body weight, which started as early as 3 days after the treatment (supplemental Figure 3). Moreover, prolonged treatment with higher doses of either imetelstat or MM (30 mg/kg) was associated with a significant reduction in spleen weights at the time the mice were euthanized as compared with the spleens from mice treated with vehicle alone (supplemental Figure 5). In order to limit the effects of imetelstat on the normal HSCs and the adverse effects on the recipient mice observed with the higher doses of the drug, we chose the 4 weeks, 15 mg/kg dose of imetelstat to treat mice transplanted with MF CD34+ cells.
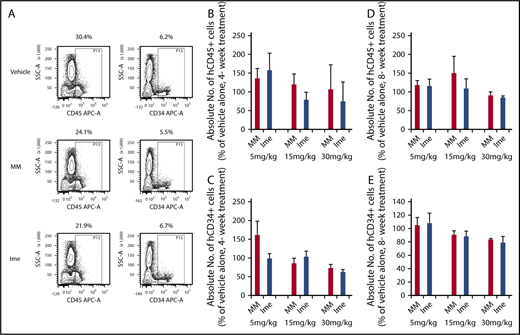
Imetelstat treatment has modest effects on normal SRCs. (A) Representative fluorescence-activated cell sorter (FACS) plots showing hCD45 cell chimerism and hCD34+ cells generated in the marrow of NSG mice receiving the same CB CD34+ cells treated with vehicle alone, MM, or Ime (15 mg/kg). (B-E) The absolute number of hCD45+ and hCD34+ cells generated in the marrow of NSG mice 4 months after the transplantation of normal CB CD34+ cells. One week after the transplantation, the recipient mice were treated with vehicle alone, MM (5-30 mg/kg), or Ime (5-30 mg/kg) for 4 weeks (B-C) or 8 weeks (D-E). The absolute number of hCD45+ and hCD34+ cells was calculated by multiplying the total number of marrow cells (cells harvested from 2 tibias, 2 femurs and 2 humeri) by the percent of hCD45+ and hCD34+ cells, respectively. The data were presented as the percentage of the absolute number of hCD45+ and hCD34+ cells present in the marrow of recipient mice treated with MM or Ime relative to that detected in the mice treated with vehicle alone. Three samples of pooled CD34+ cells from 8 to 10 CB donors were transplanted. P all > .05, MM vs Ime at each dose. Two independent experiments were performed for each sample.
Imetelstat treatment has modest effects on normal SRCs. (A) Representative fluorescence-activated cell sorter (FACS) plots showing hCD45 cell chimerism and hCD34+ cells generated in the marrow of NSG mice receiving the same CB CD34+ cells treated with vehicle alone, MM, or Ime (15 mg/kg). (B-E) The absolute number of hCD45+ and hCD34+ cells generated in the marrow of NSG mice 4 months after the transplantation of normal CB CD34+ cells. One week after the transplantation, the recipient mice were treated with vehicle alone, MM (5-30 mg/kg), or Ime (5-30 mg/kg) for 4 weeks (B-C) or 8 weeks (D-E). The absolute number of hCD45+ and hCD34+ cells was calculated by multiplying the total number of marrow cells (cells harvested from 2 tibias, 2 femurs and 2 humeri) by the percent of hCD45+ and hCD34+ cells, respectively. The data were presented as the percentage of the absolute number of hCD45+ and hCD34+ cells present in the marrow of recipient mice treated with MM or Ime relative to that detected in the mice treated with vehicle alone. Three samples of pooled CD34+ cells from 8 to 10 CB donors were transplanted. P all > .05, MM vs Ime at each dose. Two independent experiments were performed for each sample.
Inhibitory effects of imetelstat treatment on MF SRCs
We next examined the effect of treatment with imetelstat at the dose of 15 mg/kg on MF HSCs for 4 weeks. As shown in Figure 4A, 3 months after the administration of imetelstat, the degree of hCD45+ cell chimerism in the marrow of recipient mice transplanted with splenic CD34+ cells isolated from Pt 5 was reduced as compared with treatment with MM or vehicle alone. Moreover, depletion of hCD34+ cell numbers was also achieved with imetelstat treatment (Figure 4A). As shown in Figure 4B, imetelstat treatment resulted in a significant reduction in the absolute number of hCD45+ cells in the marrow of mice transplanted with cells from each of 3 patients studied (P < .05, Ime vs MM; P < .001, Ime vs vehicle alone). A reduction in the absolute number of hCD34+ cells in transplanted mice treated with imetelstat was also observed (Figure 4C; P = .14, Ime vs MM; P < .001, Ime vs vehicle alone). Such treatment only resulted in a modest reduction in body weight of mice receiving splenic CD34+ cells in 1 of the 3 patients studied (supplemental Figure 4). Furthermore, only BMCs from primary recipient mice transplanted with the same MF grafts that were treated alone with vehicle alone or MM but not imetelstat were capable of reconstituting donor derived hematopoiesis in the secondary recipient NSG mice (Figure 4D-E), indicating that that MF HSCs were eliminated for a sustained period of time by in vivo imetelstat treatment. Furthermore, imetelstat treatment decreased the JAK2V617F allele burden of hCD45+ cells isolated from the marrow of primary recipient mice receiving grafts from Pt 8 who was JAK2V617F+ (JAK2V617F allele burden: vehicle alone: 95.3%, MM: 68.9%, Imetelstat: 43.3%). These data suggest that imetelstat is capable of selectively eliminating MF HSCs but sparing their normal counterpart. In the other 2 patients studied there were not identifiable mutations available to assess the effects of imetelstat on the mutant allele burden.
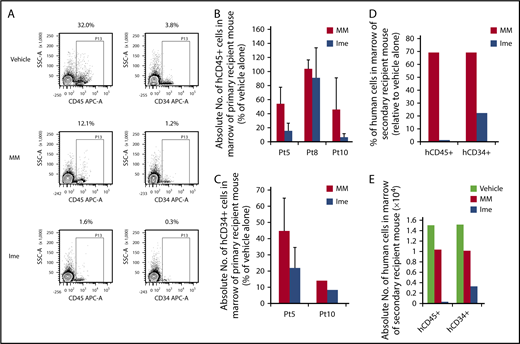
Imetelstat is capable of eliminating MF SRCs. (A) Representative FACS plots showing hCD45 cell chimerism and hCD34+ cells detected in the marrow of NSG mice receiving the same MF splenic CD34+ cells treated with vehicle alone, MM, or Ime (15 mg/kg). (B-C) The absolute number of hCD45+ and hCD34+ cells generated in the marrow of NSG mice 4 months after the transplantation of MF splenic CD34+ cells. One week after the transplantation of MF splenic grafts, the recipient mice were treated with vehicle alone or both drugs, and the data were analyzed as described in Figure 3. n = 3. HCD45+ cells: P = .03, MM vs Ime. HCD34+ cells: P = .14, MM vs Ime. Data of 3 patient samples were pooled together and analyzed. Two independent experiments were performed for each patient sample. (D) The percentage of the absolute number of hCD45+ and hCD34+ cells detected in the marrow of secondary recipient mice receiving the BMCs harvested from primary recipient mice receiving Pt 5 splenic CD34+ cells treated with MM or Ime (15 mg/kg) relative to mice receiving same graft but treated with vehicle alone. (E) Absolute number of hCD45+ and hCD34+ cells generated in the marrow of the secondary recipient mice. Half of the BMCs collected from each primary recipient mouse receiving grafts from Pt5 were injected via the tail vein into a secondary recipient mouse and BMCs from the secondary recipient mouse were then harvested 2 months after the transplantation. Only BMCs from primary recipient mice transplanted with grafts from Pt5 that were treated alone with vehicle alone or MM but not imetelstat were able to reconstitute the secondary recipient NSG mice. BMCs, BM cells.
Imetelstat is capable of eliminating MF SRCs. (A) Representative FACS plots showing hCD45 cell chimerism and hCD34+ cells detected in the marrow of NSG mice receiving the same MF splenic CD34+ cells treated with vehicle alone, MM, or Ime (15 mg/kg). (B-C) The absolute number of hCD45+ and hCD34+ cells generated in the marrow of NSG mice 4 months after the transplantation of MF splenic CD34+ cells. One week after the transplantation of MF splenic grafts, the recipient mice were treated with vehicle alone or both drugs, and the data were analyzed as described in Figure 3. n = 3. HCD45+ cells: P = .03, MM vs Ime. HCD34+ cells: P = .14, MM vs Ime. Data of 3 patient samples were pooled together and analyzed. Two independent experiments were performed for each patient sample. (D) The percentage of the absolute number of hCD45+ and hCD34+ cells detected in the marrow of secondary recipient mice receiving the BMCs harvested from primary recipient mice receiving Pt 5 splenic CD34+ cells treated with MM or Ime (15 mg/kg) relative to mice receiving same graft but treated with vehicle alone. (E) Absolute number of hCD45+ and hCD34+ cells generated in the marrow of the secondary recipient mice. Half of the BMCs collected from each primary recipient mouse receiving grafts from Pt5 were injected via the tail vein into a secondary recipient mouse and BMCs from the secondary recipient mouse were then harvested 2 months after the transplantation. Only BMCs from primary recipient mice transplanted with grafts from Pt5 that were treated alone with vehicle alone or MM but not imetelstat were able to reconstitute the secondary recipient NSG mice. BMCs, BM cells.
Mechanisms underlying the inhibitory effects of imetelstat treatment on MF CD34+ cells
Telomerase expression and activity of normal and splenic MF CD34+ cells.
Because the proposed direct cellular target of imetelstat is telomerase, we assessed if on-target effects were actually responsible for the observed effects of imetelstat on MF HSCs/HPCs. As shown in Figure 5A-B, hTERT was expressed at similar levels in primary MF splenic and normal CD34+ cells. As the enzymatic activity of telomerase depends on both hTERT and its RNA template component, we used a PCR-based assay to measure TA in primary MF splenic and normal CD34+ cells. A difference in TA between MF and normal CD34+ cells was not observed (Figure 5C). However, telomere length analysis using Flow-FISH revealed that normal HSCs (CD34+CD38−) and more differentiated progenitors (CD34+CD38+) had longer telomeres than the corresponding MF cell populations (Figure 5D). We next evaluated TA present in MF and normal CD34+ cells following exposure to imetelstat or MM and showed that treatment with imetelstat alone resulted in a significant decrease in TA in MF (Figure 5E) but not normal CD34+ cells (Figure 5F) relative to treatment with MM, indicating that the inhibitory effect of imetelstat on MF HSCs/HPCs was mediated by the inhibition of TA.
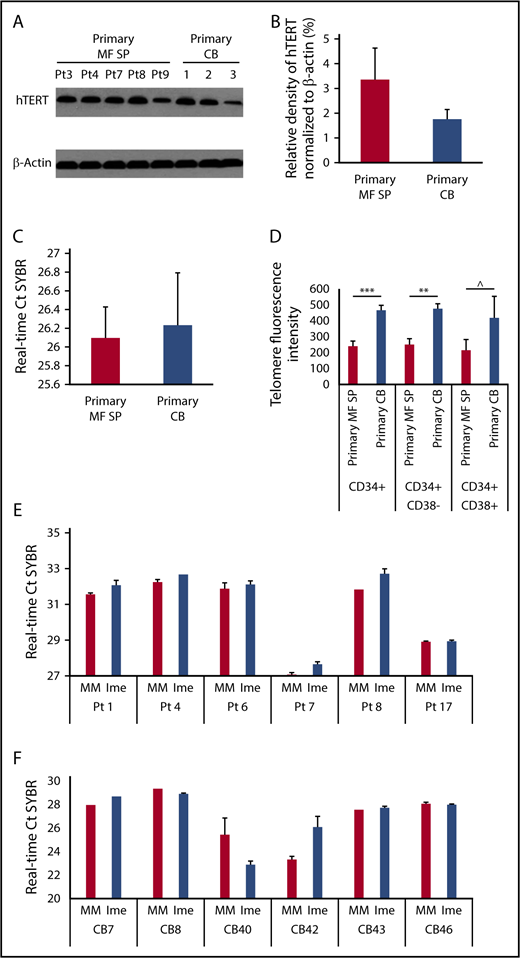
Telomerase expression and activity of normal and splenic MF CD34+cells. (A) The protein level of hTERT in primary splenic MF and normal CD34+ cells was determined by western blotting with an antibody raised against the hTERT. (B) Densitometric analysis of western blots as represented by panel A shows a similar level of hTERT in primary MF splenic and normal CD34+ cells. MF: n = 5; CB: n = 3. P > .05, MF vs CB. (C) RT-PCR–based assay for TA in primary splenic MF and normal CB CD34+ cells. The higher the Ct value indicates the lower the TA. MF: n = 8; CB: n = 3. P > .05, MF vs CB. (D) Flow-FISH analysis of telomere length of primary splenic MF and normal CB CD34+, CD34+CD38− and CD34+CD38+ cells. The higher telomere fluorescence intensity, the longer telomere. MF: n = 7; CB: n = 4. ***P = .001, **P < .01, ^P = .19. (E-F) RT-PCR–based assay for TA in splenic MF (E) and normal (F) CD34+ cells following the treatment with MM or Ime (7.5 μM) for 7 days. The treatment of MF splenic but not CB CD34+ cells with Ime resulted in a decrease in TA as compared with the treatment with MM. MF and CB: n both = 6. MF: P < .05; CB: P = .87, MM vs Ime.
Telomerase expression and activity of normal and splenic MF CD34+cells. (A) The protein level of hTERT in primary splenic MF and normal CD34+ cells was determined by western blotting with an antibody raised against the hTERT. (B) Densitometric analysis of western blots as represented by panel A shows a similar level of hTERT in primary MF splenic and normal CD34+ cells. MF: n = 5; CB: n = 3. P > .05, MF vs CB. (C) RT-PCR–based assay for TA in primary splenic MF and normal CB CD34+ cells. The higher the Ct value indicates the lower the TA. MF: n = 8; CB: n = 3. P > .05, MF vs CB. (D) Flow-FISH analysis of telomere length of primary splenic MF and normal CB CD34+, CD34+CD38− and CD34+CD38+ cells. The higher telomere fluorescence intensity, the longer telomere. MF: n = 7; CB: n = 4. ***P = .001, **P < .01, ^P = .19. (E-F) RT-PCR–based assay for TA in splenic MF (E) and normal (F) CD34+ cells following the treatment with MM or Ime (7.5 μM) for 7 days. The treatment of MF splenic but not CB CD34+ cells with Ime resulted in a decrease in TA as compared with the treatment with MM. MF and CB: n both = 6. MF: P < .05; CB: P = .87, MM vs Ime.
Imetelstat induces apoptosis of MF but not normal CD34+ cells.
We next evaluated the functional consequences of telomerase inhibition in MF CD34+ cells. As shown in Figure 6B-C, imetelstat treatment did not induce apoptosis of CB CD34+ cells. By contrast, as shown in Figure 6E, 2 days after the treatment of MF CD34+ cells with the same dose of imetelstat, the percentage of MF CD34+ annexin V+ PI− cells was significantly greater than the percentage of cells treated with MM or cytokines alone (P both <.01). The absolute number of CD34+ annexin V+ PI− cells present in imetelstat containing cultures was 3.4 ± 1.0- and 5 ± 1.4-fold greater than in cultures treated with MM or cytokines alone, respectively (Figure 6F).
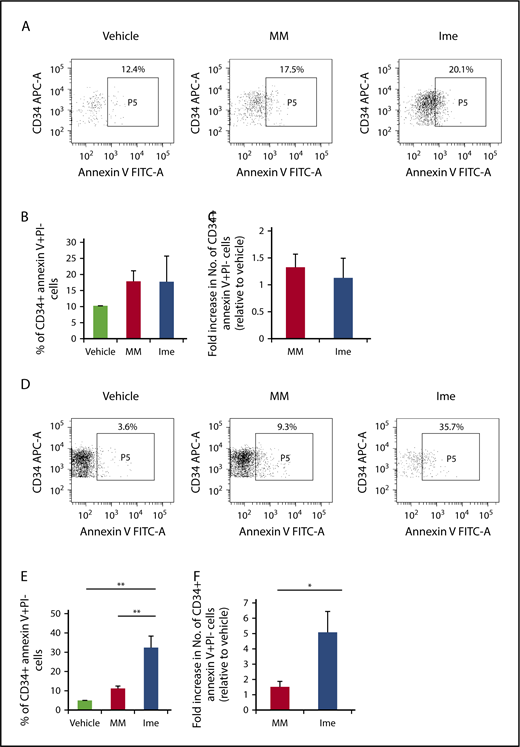
Imetelstat induces apoptosis of MF but not normal CD34+cells. (A,D) Representative FACS plots showing the percentage of CB (A) or MF (D) CD34+ cells that were undergoing apoptosis 2 days after the treatment with vehicle alone, MM, or Ime (7.5 μM). Both the percentage (B) and absolute number (C) of normal CB CD34+ cells that were annexin V+ and propidium iodide (PI)− were similar in cultures under each treatment condition. However, both the percentage (E) and absolute number (F) of MF splenic CD34+ cells that were annexin V+ and PI− were greater in cultures treated with cytokines plus Ime (7.5 μM) as compared with cells treated with cytokines plus MM (7.5 μM). *P < .05; **P < .01. MF and CB: n each = 4.
Imetelstat induces apoptosis of MF but not normal CD34+cells. (A,D) Representative FACS plots showing the percentage of CB (A) or MF (D) CD34+ cells that were undergoing apoptosis 2 days after the treatment with vehicle alone, MM, or Ime (7.5 μM). Both the percentage (B) and absolute number (C) of normal CB CD34+ cells that were annexin V+ and propidium iodide (PI)− were similar in cultures under each treatment condition. However, both the percentage (E) and absolute number (F) of MF splenic CD34+ cells that were annexin V+ and PI− were greater in cultures treated with cytokines plus Ime (7.5 μM) as compared with cells treated with cytokines plus MM (7.5 μM). *P < .05; **P < .01. MF and CB: n each = 4.
Discussion
Small-molecule inhibitors of JAK1/2 are routinely used to treat MF patients, resulting in dramatic improvement in systemic symptoms and reduction in the degree of splenomegaly without significantly affecting the JAK2V617 allele burden, BM histology, or reducing the risk of transformation to acute myelogenous leukemia (AML).38,39 Several JAK 1/2 inhibitors have been reported to be capable of inhibiting but not eliminating JAK2V617F+ HPCs.32,40 We have also reported that JAK2 inhibitors only affect a subpopulation of MF HPCs, while sparing MF HSCs.32 The inability of such agents to affect malignant HSCs likely explains why the use of JAK1/2 inhibitors is associated with disease palliation.
Recently, clinical studies of imetelstat have demonstrated possible disease-modifying activity in a subset of patients with MF.25 Using in vitro HPC assays and in vivo HSC assays, we have examined the effects of imetelstat on primary MF HSCs/HPCs. Treatment with imetelstat resulted in the depletion of MF myeloid progenitors but also selectively affected the malignant SRCs irrespective of their mutational status. By contrast, treatment of normal CD34+ cells with imetelstat in an identical fashion did not lead to inhibition of normal hematopoiesis. These data suggest that imetelstat might be capable of selectively depleting malignant MF HSCs/HPCs and sparing the reservoir of normal HSCs present in these MF spleens. Therefore, the clinical beneficial effects of imetelstat observed in the published clinical trials might be because of the differential sensitivity between MF and normal HSCs/HPCs. A similar therapeutic window for imetelstat has also been recently demonstrated by Bruedigam et al using AML xenograft models in which robust efficacy of imetelstat against AML cells was documented while normal myeloid and HSCs were largely spared.19
Imetelstat treatments have resulted in clinical and molecular responses in a subset of ET and MF patients.24,25 However, in these 2 clinical trials, it was noted that initial telomere length was not predictive of clinical responses, and that significant changes were not observed between baseline and posttreatment telomere length among patients with MF who had a clinical response, suggesting that telomere shortening was not the primary mechanism underlying the beneficial effects of imetelstat. Moreover, both studies documented clinically significant adverse effects associated with imetelstat therapy including hepatotoxicity, myelosuppression, and bleeding tendencies. Thrombocytopenia has been one of the major dose-limiting toxicities associated with imetelstat administration.21,25,41 Many have suggested that imetelstat’s actions in MF and ET patients were because of off-target effects.42 In order to more precisely understand the drug’s mechanism of action, we performed in vitro and in vivo assays with parallel treatments including imetelstat, vehicle alone, and MM, which possesses the same lipid-conjugated thiophosphoramidate chemistry as imetelstat but is unable to hybridize to hTR. Such comparison treatments are not possible to perform during clinical trials involving patients. We, however, did observe that treatment with MM at a dose and schedule used that did not inhibit normal human SRCs did reduce MF SRCs by 50% with CD34+ cells from 2 of 3 different MF patients studied (Figures 3 and 4). Using a patient-derived xenograft (PDX) model, treatment with imetelstat led to greater depletion or elimination of MF SRCs but not their normal counterparts (Figure 4B-C), and imetelstat was capable of eliminating MF HSCs that were capable of repopulating secondary hosts (Figure 4D-E). Notably, these observations in the MF PDX model were made 3 months after the imetelstat treatment had been discontinued. These data indicate that imetelstat had a profound and sustained effect on MF HSCs. Furthermore, we observed that imetelstat suppressed TA of MF CD34+ cells and induced greater degrees of their apoptosis. In addition, the telomere length of MF stem and progenitor cells was shorter (2×) as compared with their normal counterparts, which might further increase their sensitivity to imetelstat treatment. Taken together, these results provide direct evidence that inhibitory actions of imetelstat observed in vivo in MF HSCs/HPCs are attributable at least in part to on-target effects.
Our in vitro and in vivo studies have also demonstrated that the inhibitory effects of imetelstat on MF HSCs/HPCs occurred irrespective of the patient’s driver mutation. However, recent clinical studies have indicated that responses occurred in 27% of MF patients with a JAK2 mutation but not in patients that lacked this mutation.25 By contrast, Baerlocher et al revealed that imetelstat therapy was capable of reducing the driver mutation allele burden of ET patients when these patients were treated in a clinical trial. Our findings suggest that imetelstat was active in MF patients irrespective of the driver mutational status (supplemental Figure 1; Figure 4B-C). Clearly limited numbers of patients were evaluated in this study and the clinical trials conducted by Tefferi et al and Baerlocher et al. Larger numbers of patients with different driver mutations should be studied to resolve this issue. The use of a PDX model to study the effects of MF HSCs depleting agents appears to be a valuable method with which to evaluate the effects of therapeutic agents against MF and normal hematopoiesis.
Our in vivo studies demonstrated that reduced numbers of normal hCD45+CD41a+ mature megakaryocytes were observed in mice transplanted with CB CD34+ cells from 2 out of 3 pooled samples transplanted 3 months after the discontinuation of imetelstat treatment (30 mg/kg) (supplemental Figure 2), whereas normal HSCs/HPCs were modestly affected (Figure 3B-C). Although the inhibitory effect of imetelstat on MF megakaryogenesis was not able to be evaluated because no hCD45+CD41a+ cells were generated in mice transplanted with splenic CD34+ cells from any of the 3 MF patients studied, our findings from the in vivo studies with normal CD34+ cells suggest that the high doses of imetelstat might affect platelet biogenesis, which would eventually lead to thrombocytopenia in patients. These findings are consistent with those previously reported by Iancu-Rubin et al. Using a well-established system of ex vivo megakaryopoiesis, they demonstrated that imetelestat treatment affected normal megakaryocyte development by exclusively delaying maturation of MK precursor cells.43
In conclusion, imetelstat is capable of selectively depleting malignant HSCs/HPCs from patients with MF by inhibiting TA and inducing apoptosis, which might account for the clinical benefits reported in the phase 1/2 studies in MPN patients. What is remarkable about our findings is the persistence of the depletion of the MF HSCs for at least 3 months after therapy was completed. A phase 2 trial of imetelstat therapy in MF patients has been performed at multiple institutions, and the outcome of this study is eagerly awaited. Clearly from our in vivo studies, the intensity of the schedule of administration plays a major determinant role in its efficacy and possible toxicity. Further attention to the dose and schedule of administration of imetelstat might be needed in order to translate the effects of depleting MF HSCs that we have demonstrated to a clinical trial.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the Center for Comparative Medicine and Surgery of ISMMS for their help with mouse BM section preparation and hematoxylin and eosin staining.
This work was supported by Geron Corporation and Janssen Research & Development LLC. X.W. has received research funding for this work from Geron Corporation and Janssen Research & Development LLC.
Authorship
Contribution: X.W. designed the study, performed the experiments, interpreted the data, and wrote the manuscript; C.S.H. performed most of the experiments and analyzed the data; B.P. interpreted histopathology; J.Q. performed part of the experiments; F.Y. and J.H. performed JAK2 mutational analysis; K.E. and F.H. analyzed data and discussed results; and R.H. interpreted the data and revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Xiaoli Wang, Division of Hematology/Medical Oncology, Department of Medicine, The Tisch Cancer Institute, Myeloproliferative Neoplasms Research Consortium, Icahn School of Medicine at Mount Sinai, 1 Gustave L. Levy Pl, Box 1079, New York, NY 10029; e-mail: xiaoli.wang@mssm.edu.



