Key Points
We have identified hierarchal subtype differences in myeloma cells at diagnosis.
The prognostic impact supports an acquired B-cell trait and phenotypic plasticity as a pathogenetic hallmark of MM.

Abstract
Despite the recent progress in treatment of multiple myeloma (MM), it is still an incurable malignant disease, and we are therefore in need of new risk stratification tools that can help us to understand the disease and optimize therapy. Here we propose a new subtyping of myeloma plasma cells (PCs) from diagnostic samples, assigned by normal B-cell subset associated gene signatures (BAGS). For this purpose, we combined fluorescence-activated cell sorting and gene expression profiles from normal bone marrow (BM) Pre-BI, Pre-BII, immature, naïve, memory, and PC subsets to generate BAGS for assignment of normal BM subtypes in diagnostic samples. The impact of the subtypes was analyzed in 8 available data sets from 1772 patients’ myeloma PC samples. The resulting tumor assignments in available clinical data sets exhibited similar BAGS subtype frequencies in 4 cohorts from de novo MM patients across 1296 individual cases. The BAGS subtypes were significantly associated with progression-free and overall survival in a meta-analysis of 916 patients from 3 prospective clinical trials. The major impact was observed within the Pre-BII and memory subtypes, which had a significantly inferior prognosis compared with other subtypes. A multiple Cox proportional hazard analysis documented that BAGS subtypes added significant, independent prognostic information to the translocations and cyclin D classification. BAGS subtype analysis of patient cases identified transcriptional differences, including a number of differentially spliced genes. We identified subtype differences in myeloma at diagnosis, with prognostic impact and predictive potential, supporting an acquired B-cell trait and phenotypic plasticity as a pathogenetic hallmark of MM.
Introduction
Despite the extensive insight into multiple myeloma (MM) pathogenesis, as outlined in the World Health Organization classification,1,2 a number of questions remain unanswered regarding the origin and initiation of the developing myeloma cells, including its association with the normal B-cell hierarchy in the bone marrow (BM).3-6 We hypothesize that considering MM as a disease of differentiation by identifying its cell of origin (COO) could lead to novel biological insight and development of new treatment options as described by Boise et al.7
MM develops from a premalignant monoclonal gammopathy of unknown significance (MGUS), by a stepwise oncogenesis to intramedullary early smoldering or evolving de novo myeloma because of acquired genetic deregulation.8-10 The primary translocations implicating the 14q32 locus involve a series of promiscuous target genes, with CCND1 and FGFR3/MMSET being the most frequently present at the MGUS stage.11 Furthermore, the larger part of break points occurs in the switch regions, suggesting the early translocation happens during immunoglobulin heavy chain class-switch recombination in the germinal center.12-14 The existence of early translocations and the overexpression of CCND genes form the translocations and cyclin D (TC) classification generated from early events.9,11 Later incidences include a spectrum of mutations and dysregulations occurring in advanced disease with poor prognosis.14-19
Myeloma plasma cells (PCs) are class switched, freezing the initiating cell at the postgerminal B-cell maturation stage, refuting that the disease is initiated in earlier B-cell subsets, as has been proposed before.20 The earliest immunoglobulin heavy chain clonotypic cell we have identified with a class-switched isotype is in the myeloma memory B-cell compartment,21,22 but its clonogenic and malignant potential is a controversial issue.23-26 Recent studies have concluded that the clonotypic cells are remnants of a neoplastic cell with no malignant potential,27,28 contrasting the myeloma PC compartments.
The myeloma stem cell concept has been reviewed in detail by us and others.29,30 We proposed an operational definition of COO to allow for acquisition of data supporting that a normal B cell, which achieves the first myeloma initiating mutation, is not necessarily linearly connected to the myeloma stem cell. These results underpin the hypothesis that myeloma generating cells are present in the malignant PC compartment, but the COO is a normal counterpart of a germinal-center B cell that evolves via differentiation into a premalignant PC compartment already present in MGUS populations.
The plasticity potential of myeloma cells, perhaps caused by interaction with the tumor microenvironment, also plays an important role in development and maintenance of MM.30
The present study takes a COO approach, where we refer to an expanding compartment initiated by a differentiation specific oncogene hit.31 The terms “COO” and “cancer stem cells” have been used interchangeably. However, it is important to differentiate between them as in contrast to our phenotypic COO studies, it is our perception that cancer stem cell research depends on single cell studies in the frame of the classical stem cell definition.29
The deregulated B-cell cells under influence by the microenvironment may be key in the emergence of myeloma and its related phenotypic changes. This phenomenon, coined “plasticity,” is defined as a changed cellular phenotype or function during deregulated differentiation.32 More specifically, this refers to malignant mature PCs that share properties of different maturation steps, including precursors. The phenomenon facilitates a new tool for providing insight into the observed clonal plasticity33,34 associated with oncogenesis.8-12,17,35-38 The mechanisms of deregulated differentiation and myeloma-cell plasticity ought to be investigated and their clinical significance assessed.
Recently, we have documented a procedure to identify and study gene expression of flow-sorted human B-cell subsets from normal lymphoid tissue.39-42 These subsets can be profiled and by proper statistical modeling defined specific B-cell associated gene signatures (BAGS), recently introduced for diffuse large B-cell lymphoma (DLBCL).43-45 Here we have applied BAGS from normal BM subsets to assign individual MM subtypes and correlate them with prognosis to delineate their pathogenetic impact.
Patients, material, and methods
The subtyping method based on normal BM has previously been briefly outlined in Nørgaard et al46 and applied to chronic lymphocytic leukemia patients. In this article, we will describe it in more detail and provide more profound quality control of the normal samples.
Ethical statement and tissue collection
All normal tissue samples were collected in accordance with the research protocol (Myeloma Stem Cell Network, N-20080062MCH) accepted by the North Denmark Regional Committee on Health Research Ethics.
Normal BM was either harvested from the sternum (n = 7)41 during cardiac surgery or taken as aspirates from the iliac crest of healthy volunteers (n = 14) during routine surgical procedures in accordance with the Declaration of Helsinki. Normal BM aspirates were cleared for red blood cells by 15 minutes lysis using 20-fold excess of Easylyse (DAKO, Glostrup, Denmark). The sample was centrifuged 10 minutes at 250g, the supernatant discarded, and the pellet washed in phosphate-buffered saline. The centrifugation step was repeated, and the pelleted cells dissolved in stain buffer and processed directly for analysis using multiparametric flow cytometry (MFC).
MFC, cell sorting, and gene expression profiling
The normal B-cell subsets were phenotyped by MFC and fluorescence-activated cell sorting (FACS) into 6 distinct B-cell subsets (Pre-BI, Pre-BII, immature [Im], naïve [N], memory [M] B cells, and PCs) using a monoclonal antibody panel,41,44 described in detail in supplemental Text 1.
RNA was labeled for microarray analysis, hybridized to the Affymetrix Human Exon 1.0 ST (HuEx-1.0-st) Array41,44 or to the Affymetrix Human Genome U133 Plus 2.0 (HU-U133-2.0) Array platform, and referred to as the “normal sternal BM data set” (n = 7) (GSE68878),47 the “normal iliac crest BM aspirate” data set–HuEx-1.0-st (n = 8) (GSE99635), and the “normal iliac crest BM aspirate” data set–Hu-U133-2.0 (n = 6) (GSE107843). The CEL files have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus repository with GSE numbers as indicated previously and comply with minimum information about a microarray experiment (MIAME) requirements.48
Clinical myeloma data sets
Clinical data originated from Affymetrix microarray analysis of PC-enriched myeloma samples, from 4 prospective clinical trials: University of Arkansas for Medical Sciences (UAMS), Hematology Oncology Group-65/German-speaking Myeloma Multicenter Group-HD4, Medical Research Council Myeloma IX (All HG-U133-2.0 arrays), and Acute Medically Ill Venous Thromboembolism Prevention with Extended Duration Betrixaban (APEX) (Affymetrix Human Genome U133A [HG-U133A] arrays),35,36,49-52 as well as a preclinical study, the Intergroupe Francophone du Myelome and Dana-Farber Cancer Institute Project37,53 (HuEx-1.0-st arrays).
The following 3 cohorts were used for BAGS assignment and subtype association in the disease stages MGUS, smoldering multiple myeloma, relapsed multiple myeloma, primary plasma cell leukemia, and human myeloma cell lines: the Salamanca cohort (Affymetrix Human Gene 1.0 ST arrays),54 the Milan cohort (HG-U133A arrays),55 and the Mayo cohort (HG-U133A arrays).56,57
The following 2 B-cell line data sets were used in the analysis: Bcell20 (HuEx-1.0-st arrays) and Bcell26 (HG-U133-2.0 arrays).58
All data sets are retrieved from Gene Expression Omnibus with the indicated GSE numbers and are described in detail in the “Materials and methods” of supplemental Text 1.
Statistical analysis
Array normalization.
All gene expression data sets where cohort-wise background corrected, normalized, and summarized by robust multichip average (RMA)62 normalization using the functions from the R-package affy v1.54.063 and summarized at Ensemble Gene ID level with a custom CDF file from brainarray version 20.0.0. The analyses are based on the online gene expression profile (GEP) data sets listed previously and summarized in supplemental Text 1.
Systematic evaluation of transcription factors, surface markers, and B-cell differentiation genes.
Transcription factors, surface markers, and B-cell differentiation genes as defined in Biomart (http://www.biomart.org/) were listed and included in hierarchical clustering of the BM data set. The most variable and distinctive genes across subsets were selected and combined with B-cell subset specific genes identified through literature review to evaluate subset identity.
Multivariate statistical methods.
Comparisons between GEPs of molecular subsets were performed by principal component analysis and heat maps. For this we used the prcomp and heatmap.2 functions from the R-packages stats and gplots, respectively. The hierarchical clusters alongside the heat maps were made using Ward’s method with Pearson’s correlation coefficient as dissimilarity measure via the R-function hclust.
TC and risk classification.
Methods for deriving the TC expression pattern and the UAMS risk classification based on Affymetrix gene expression microarray data are described in Bergsagel et al and Zhan et al.9,35
BAGS classification.
The BAGS classifier was trained on median centered gene expression data from normal BM, using a regularized multinomial regression model with 6 classes corresponding to the respective B-cell subtypes. Regularization was done using the elastic net penalty as implemented in the R-package glmnet.64 The elastic net penalty depends on a model parameter α varying between 0 and 1, which balances the ridge regression penalty (α = 0), allowing all genes in the model, and the Lasso penalty (α = 1), allowing a maximum number of genes equal to the number of samples. The model also involves a regularization parameter λ that determines the weight of the penalty and thereby the number of nonzero coefficients. The model parameter α was varied between 0.01 and 1, and the regularization parameter λ was varied between −7.5 and −1 on a log scale. The adequacies of the models were validated by choosing the minimum multinomial deviance determined by “leave 1 sample out” cross validation. The model with α = 0.24 and log λ = −5.67 had the smallest cross-validated multinomial deviance leaving us with 184 genes with nonzero coefficients (supplemental Figure 1). This indicates, that by lowering log λ below −5.67, a number of alternative very gene rich models could have described the data well. However, we found the above-mentioned choice a suitable compromise between sparsity and robustness. Each clinical data set was gene-set-wise adjusted to have zero median and same variance as the normal BM data set. The associated COO Pre-BI, Pre-BII, Im, N, M, or PC for each patient in each data set was subtyped by the BAGS classifier by assigning the class with the highest predicted probability score above 0.40 and otherwise unclassified (supplemental Figure 2).
The probability cutoff was thoroughly tested (supplemental Text 2, section 11.1).
REGS classification.
Survival analysis.
Kaplan-Meier curves, log-rank tests, and Cox proportional hazards models were used for survival analysis and handled with the R-package survival.69 The cohorts involving patients from 3 prospective clinical trials were amalgamated into a meta–data set to increase the power of the study. BAGS subtypes as an independent explanatory variable was investigated in the meta–data set by a Cox proportional hazards regression analysis with BAGS subtypes, TC classes, and cohort as potential confounders. Harell’s C-statistic for overall survival (OS) was calculated from predicted values from the multivariate Cox model with and without inclusion of the BAGS classes to assess the prognostic utility.
Detection of alternative splice variants in BAGS subclasses.
Alternative splice variants in the BAGS subtypes were investigated using the analysis of splice variation (ANOSVA) method.70 The method consists of a linear model with interaction effects to detect alternative exon usage between 1 class and the rest within a gene. The linear model is given by: yijk = μ + αi + βj + γij + εijk, where yijk is the log2 RMA normalized gene expression for class i, exon j, and sample k; μ is the mean expression for the gene; αi is the class effect; βj is the exon effect; γij is the interaction between group i and exon j (ie, the alternative exon usage); and the εijk’s are independent, identically distributed Gaussian residuals. The analysis was performed genewise for all genes of interest, and the result for each gene was reported as the effect size and P value for the most significant interaction effect. When fitting a linear model in R, the interaction effects are by default given as treatment effects (ie, 1 level is set as a reference with an effect of zero and other effects are estimated as deviations from this). This makes the estimation dependent on the choice of reference. As we are interested in deviations from the group effect, an internal loop over exons was initially run for each gene, where the expressions were groupwise median centered and the reference exon was chosen as the exon giving the highest P value in a 2-sided Student t test of the group means (ie, the exon where the difference between groups was closest to the median difference). For the analysis of differential exon usage, the uncentered data were used.
Data for the analysis were obtained by RMA normalizing the 170 samples with HuEx-1.0-st array data from the IFM-DFCI data set using the just.rma function from the R-package affy v1.54.063 and summarizing at exon level with a custom CDF file from brainarray version 20.0.0. All genes included in the training set for the BAGS classifier were investigated for differential exon usage for Pre-BII vs rest, and memory vs rest. Genes with evidence of differential exon usage were defined as genes with an adjusted P < .01 and an absolute interaction effect >1. Significant genes for the analysis of memory subtype cases vs rest are shown in supplemental Table 1B, and significant genes for the analysis of Pre-BII vs rest are shown in supplemental Table 2A.
An enrichment analysis for biological process gene ontology (GO) terms of significant genes was done using the R-package topGO v2.28.0. Results for memory vs rest are shown in supplemental Table 3B, and results for Pre-BII vs rest are shown in supplemental Table 3A. Updates in the GO.db database might cause changes in the enrichment analysis; accordingly, all tables and interpretations of GO terms in this document are at version 3.4.1 of the GO.db.71
The significance level was set throughout to 0.05, and effect estimates were provided with 95% confidence intervals. P values for the differential gene expression and alternative splice analyses were adjusted by Holm’s method.72
Results
BAGS classifier generation and clinical sample assignment
The data quality of the differentiating B-cell subset compartments of the sternal BM was individually validated as illustrated by density plots from MFC analysis (Figure 1A) and principal component analysis (Figure 1B) of the MFIs of the CD markers used for FACS; unsupervised cluster analysis was also conducted for the gene expression values of the membrane CD markers used for FACS (Figure 1C; previously shown in Nørgaard et al46 ) and 45 classical B-cell markers summarized from a literature search (Figure 1D). Subset-specific segregation was further documented by principal component analysis (supplemental Figure 3).
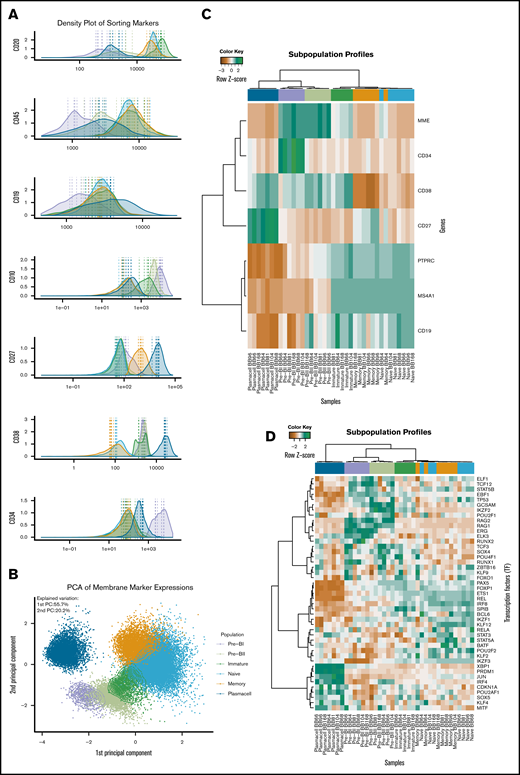
Expression of membrane markers, transcription factors, and B-cell subset-specific genes in normal BM tissue. (A) B cells of the BM were defined by flow cytometry as CD19+, CD45+, and CD3− and were additionally divided by surface marker expression of CD10, CD20, CD27, CD38, and CD34, published in detail previously.44 The data quality of the differentiating B-cell subset compartments was validated as illustrated by normalized histograms of (A) the mean fluorescence intensities (MFIs) CD markers based on merged MFC reanalysis of pure sorted populations resulting from 7 independent sorting procedures. Broken lines represent MFI values for each sorted B-cell subset. (B) Principal component analysis of the MFI values for each sorted cell in all samples. The cells are coded with a color according to their original subset. The dots represent mean values for each sorted B-cell subset. (C) The most variable probe sets were used in unsupervised hierarchical clustering analysis of the surface markers (MME = CD10, CD34, CD38, CD27; PTPRC = CD45; MS4A = CD20, CD19) used for FACS. (D) B-cell differentiation–specific genes (n = 45), summarized from a literature review of transcriptional regulation of B lymphopoiesis. The colors at the top of panel D indicate the relative gene expression for each sample, with blue representing high and brown representing low.
Expression of membrane markers, transcription factors, and B-cell subset-specific genes in normal BM tissue. (A) B cells of the BM were defined by flow cytometry as CD19+, CD45+, and CD3− and were additionally divided by surface marker expression of CD10, CD20, CD27, CD38, and CD34, published in detail previously.44 The data quality of the differentiating B-cell subset compartments was validated as illustrated by normalized histograms of (A) the mean fluorescence intensities (MFIs) CD markers based on merged MFC reanalysis of pure sorted populations resulting from 7 independent sorting procedures. Broken lines represent MFI values for each sorted B-cell subset. (B) Principal component analysis of the MFI values for each sorted cell in all samples. The cells are coded with a color according to their original subset. The dots represent mean values for each sorted B-cell subset. (C) The most variable probe sets were used in unsupervised hierarchical clustering analysis of the surface markers (MME = CD10, CD34, CD38, CD27; PTPRC = CD45; MS4A = CD20, CD19) used for FACS. (D) B-cell differentiation–specific genes (n = 45), summarized from a literature review of transcriptional regulation of B lymphopoiesis. The colors at the top of panel D indicate the relative gene expression for each sample, with blue representing high and brown representing low.
The BAGS classifiers with the smallest deviance determined by cross validation consisted of 184 genes (for details, see supplemental Text 1 and supplemental Figure 1). Each B-cell subset signature contained 27 to 54 genes, ensuring comparable gene representation for all subsets in the BAGS classifier. The selected genes and associated coefficients for the BAGS signatures are shown in supplemental Table 4 (previously shown in Nørgaard et al46 ). The expressed signatures included 51 genes associated with specific B-cell functions, 79 specific genes with more fundamental biological associations, and 24 probes with unknown gene functions (supplemental Table 5A-F).
We subsequently validated the BAGS classifier, which was trained using GEP data from Human Exon 1.0 ST Arrays, on independent normal B-cell subsets from BM aspirates from the iliac crest profiled using either HuEx-1.0-st or HG-U133-2.0 arrays, and found concordant assignments for both platforms (supplemental Table 2). This documented the cross-platform validity of the classifier, allowing its use in clinical data with GEP originating from other platforms.
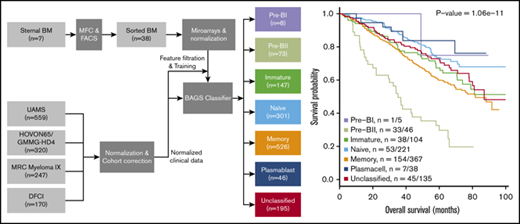
Microarray data from 4 independent cohorts (n = 1296) of de novo MM patients were assigned for BAGS subtypes. Table 1 shows the resulting assignment of the tumors and exhibited BAGS subtype frequencies and average percentage for Pre-BI = 1%, Pre-BII = 6%, Im = 11%, N = 23%, M = 41%, and PC = 4%, with no significant variation between the cohorts from different geographic regions, time periods, or sampling methods (P = .9). We allow 15% of cases within each cohort to be unclassified, resulting in a probability cutoff of ∼0.40. The distribution of the TC classes within the BAGS subtypes is given in Table 1, with significant association identified (P < .001). There was also significant correlation with ISS staging (P = .032), with increased numbers of the Pre-BII and M subtypes associated with ISS stage III, as shown in Table 1. BAGS, proliferation index, and melphalan resistance assignments for all samples used in the analyses are provided in supplemental Table 6A-H.
Prognostic impact of assigned BAGS subtypes
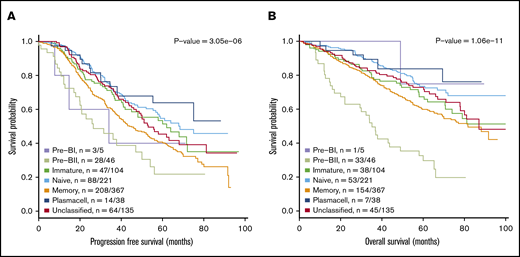
Figure 2 illustrates the results from a meta-analysis of the 916 patients included in the 3 prospective trial cohorts with high-dose melphalan as first-line therapy (UAMS, HOVON65/GMMG-HD4, and MRC Myeloma IX) with the HG-U133-2.0 microarray data available, documenting that the assigned BAGS subtypes were significantly associated with PFS and OS (PFS, log-rank test, P < .001; OS, log-rank test, P < .001). Major impact was observed within patient cohorts with the Pre-BII and M subtypes, which had a significantly inferior prognosis compared with the patients with Im, N, and PC subtypes.

Meta-analysis of the prognostic impact of assigned BAGS subtypes. Progression-free survival (PFS) (A) and OS (B) were compared between BAGS subtypes for high-dose melphalan–treated patients in published prospective clinical trials. P values are results from log-rank tests. The subtype numbers given as n are the numbers of events/number of assigned patients with the subtypes in the meta–data set. The BAGS subtypes are color coded as in Figure 1.
Meta-analysis of the prognostic impact of assigned BAGS subtypes. Progression-free survival (PFS) (A) and OS (B) were compared between BAGS subtypes for high-dose melphalan–treated patients in published prospective clinical trials. P values are results from log-rank tests. The subtype numbers given as n are the numbers of events/number of assigned patients with the subtypes in the meta–data set. The BAGS subtypes are color coded as in Figure 1.
The robustness of the BAGS association with outcome was successfully evaluated for a wide range of probability cutoffs for the percentage of unclassified cases (supplemental Text 2, section 11.1). The BAGS-assigned MM subtypes in the individual prospective clinical trial data sets UAMS/TT2&3, HOVON/GMMG-HD4, and MRCIX, all including high-dose melphalan and a variety of new drugs, were also separately analyzed for outcome following treatment as illustrated in supplemental Figure 4. Results from the individual data sets were in accordance with the above-described meta-analysis illustrated in Figure 2.
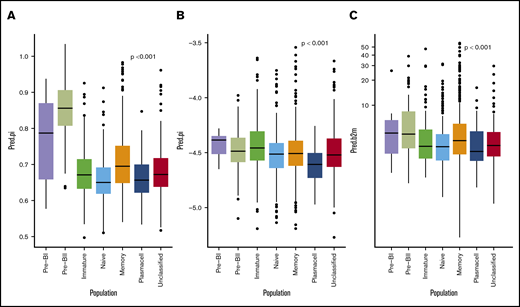
Cox proportional hazard meta-analysis results, as shown in Table 2, and Harell’s C-statistic, giving the concordance between observed survival and predicted risk scores from the a multivariate cox model with (C = 0.65) and without (C = 0.59) BAGS classes, demonstrated that the BAGS subtypes added significant and independent prognostic information to the already well-established TC classification. In addition, we found significant correlation between the BAGS subtypes and the proliferation index (PI) risk profiling (P < .001), melphalan resistance probability (P < .001), and β-2 microglobulin plasma level (P < .001) as illustrated in Figure 3, respectively. Results in these figures are for a combined data set adjusted for differences in individual data sets, while results for individual data sets may be found in supplemental Text 1 and supplemental Figure 5.
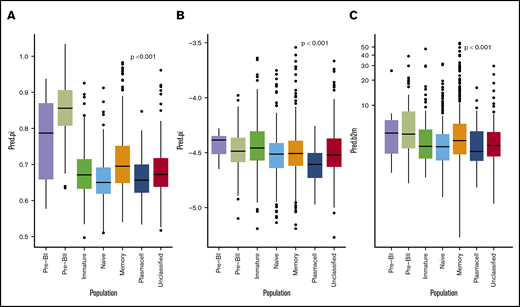
BAGS subtype boxplots with correlation to proliferation, melphalan resistance, and β-2 microglobulin. The individual adjusted PI risk profiling (A), melphalan drug resistance probability (index) (B), and β-2 microglobulin plasma level (C), respectively, per BAGS subtype cases from analysis of the meta–data set. The BAGS subtypes are color coded as in Figure 1.
BAGS subtype boxplots with correlation to proliferation, melphalan resistance, and β-2 microglobulin. The individual adjusted PI risk profiling (A), melphalan drug resistance probability (index) (B), and β-2 microglobulin plasma level (C), respectively, per BAGS subtype cases from analysis of the meta–data set. The BAGS subtypes are color coded as in Figure 1.
BAGS assignment of MGUS, smoldering myeloma, MM, extramedullary MM, and myeloma cell lines
Available data sets were used for BAGS assignment of associated myeloma diseases, as shown in supplemental Table 7A. Of interest, 5 of 6 PC leukemia cases were M subtypes, indicating a subtype evolution or selection for advanced disease. In contrast, MGUS cases had a significantly high frequency (>50%) of N subtypes, which was different from smoldering myeloma and newly diagnosed and relapsed MM. The distribution of M component isotypes showed no significant differences across BAGS subtypes, except for a tendency for light chain disease to be overrepresented in the postgerminal subtypes as shown in supplemental Table 7B. Frequencies of BAGS subtypes in relapsed MM patients from the APEX data set (supplemental Table 7C) were similar to the frequencies in first-line patients shown in Table 1. Finally, we observed that 9 out of 12 human myeloma cancer cell lines were classified as PC subtypes (supplemental Table 8).
Characterization of BAGS subtypes
Differential expression analysis of BAGS subtypes with poor prognosis (Pre-BII or memory vs rest) identified hundreds of genes with a highly significant differential expression as given in supplemental Table 9A-B for the Pre-BII and memory subtypes, respectively. GO enrichment of significant genes showed that the Pre-BII subtype myelomas are enriched for the categories mitotic cell cycle, nuclear division, and DNA-dependent DNA replication (supplemental Table 10A), and memory subtype myelomas are enriched for the categories cell-cell signaling, synaptic transmission, and multicellular organismal process (supplemental Table 10B). For more details, see supplemental Text 1.
Finally, in order to detect whether the Pre-BII and memory subtypes showed alternative splicing patterns associated with oncogenesis, we investigated alternative exon usage in the IFM-DFCI data set. Results suggested Pre-BII-specific alternative exon usage for 16 genes (supplemental Table 1A), which were especially associated with biological processes involved in cell cycle regulation (supplemental Table 3A). In memory subtype cases, we only identified 2 candidate genes with potential alternative exon usage (supplemental Table 1B) associated with basic cell functions including regulation of programmed cell death, metabolism, and signaling transduction (supplemental Table 3B). Comparison with alternative exon usage patterns detected in nonmalignant samples (supplemental Table 11) indicated that the majority of events were specific to malignant samples, suggesting association to oncogenesis.
Discussion
We have phenotyped distinct cellular subsets of B-cells in the normal BM to generate a BAGS classifier and have documented that the assigned subtypes have prognostic impact.
A probability estimate for each sample to be assigned to each of the 6 BAGS subtypes was provided. Samples with very low classification probabilities were labeled as unclassified. The frequency of unclassified samples in other gene expression–based COO classifications is ∼15%.17 A pragmatic probability cutoff of 0.40 was used, which is well above the random assignment probability of 1 out of 6, to ensure that 85% of the samples would be BAGS subtypes. The robustness of the BAGS association with outcome was successfully assessed for a wide series of probability cutoffs.
The present study was whenever possible conducted according to guidelines of –omics-directed medicine (eg, McShane et al,73 REMARK,74 and MIAME48 ). However, it is worth noting that the BAGS classifier used cohort-based normalization, which implies that it cannot practically be used in a clinical setup where patients show up 1 at a time. Remedies to this problem have been proposed elsewhere75 and were not further pursued here.
The assignment of BAGS subtypes to MM may explain an interindividual disease heterogeneity, which could reflect the association between cellular differentiation and oncogenesis.27,76-78 A standardized flow cytometry immunophenotyping of hematological malignancies illustrates the potential clinical application of surface expressed markers to identify diagnostic tumor clones.79 Such a strategy has allowed new studies of normal PC heterogeneity by differentiation.6,10,80
MM is an example of a malignant disease that has been studied intensively with microarrays. Many peer-reviewed papers have documented new classification systems based on gene expression profiles to correlate with biology and prognosis.1,8-12 The present work addresses a need to study a new diagnostic platform defined by the molecular classification of BAGS in MM, as for DLBCL.43,81 In accordance with our studies in DLBCL, where we applied the whole lymphoid differentiation compartment from tonsils or normal lymph nodes, we prospectively analyzed the lymphoid subset–defined compartments from normal BM to generate 6 BAGS for MM assignment. The idea was that the COO concept would hold true also for MM and assign subtypes from the postgerminal differentiation pathway. To our surprise, a major fraction of patient tissues were assigned a Pre-BII, Im, or N subtype, disproving the subtyping to be true reminiscence of the origin from a germinal or postgerminal phenotype. Given the phenotypic variation among MGUS, smoldering myeloma, MM, MM relapse, extramedullary MM, PC leukemia, and human myeloma cancer cell lines, it is more likely that BAGS assignment does classify MM cases based on reversible phenotypic plasticity.33
The BAGS classification is correlated to the well-established TC classification; however, we found that the poor prognosis for Pre-BII and memory subtypes correlated with the myeloma cell PI and the β-2 microglobulin plasma, but not gene expression level. PI and β-2 microglobulin is historically the most important and persistent biomarker in different trials, independent of the evolving therapy. The mechanisms behind these prognostically useful markers are unknown but should now be studied to understand their pathogenetic impact.
Our detection of alternative exon usage suggested subtype specific patterns, supporting that the BAGS phenotyping is based on biological processes. Alternatively spliced candidate genes detected in the Pre-BII subtype revealed an overrepresentation of genes involved in cell cycle regulation and increased proliferation, such as GTSE1, PKMYT1, BIRC5, and AURKB, suggesting an association with altered cell cycle regulation and proliferation. However, detected alternative exon usage of candidate genes needs to be experimentally validated and confirmed.
High-dose melphalan forms the basis of MM treatment.82 However, patients with refractory or relapsed diseases represent a large unmet need for drug-specific predictive tests and precise companion diagnostics.58,65-68 This need can be exemplified by REGS and BAGS classification with predictive information to guide therapy. The current analyses indicate that such information is available at diagnosis (Figure 3B; supplemental Figure 5B) and could be used for identification of candidates for more precise strategies. Collectively, this result indicates BAGS subtypes experience different clinical tracks and drug resistant mechanisms, and maybe even different molecular pathogenesis. We believe our results support the future inclusion of gene expression profiling in randomized prospective clinical trials aimed at improving MM treatment.
BAGS classification divided de novo MM patients into so-far-unrecognized, differentiation-dependent prognostic groups. These prognostic analyses and observations support the idea that BAGS classification in MM may contribute with pathogenetic information, especially in attempts to understand the biology behind the classical and still meaningful biomarkers PI and β-2 microglobulin. Most importantly, the classification included pregerminal subtypes, pointing at a reversible phenotypic plasticity in myeloma PCs. Prospective future studies are needed to prove the concept using clinical end points, including prediction of therapeutic outcome.
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession numbers GSE68878, GSE99635, and GSE107843).
Acknowledgments
The authors acknowledge the tremendous perseverance by the late H.E.J. in driving the project behind this publication. Unfortunately, he did not experience the final outcome of the work. Technicians Louise Hvilshøj Madsen and Helle Høholt participated in several aspects of the laboratory work.
This work was supported by research funding from the European Union Sixth Framework Programme to the Myeloma Stem Cell Network (LSHC-CT-2006-037602), the Danish Cancer Society, the Danish Research Agency to the project Chemotherapy Prediction (#2101-07-0007), and the KE Jensen Foundation (2006-2010) to H.E.J. and K.D.
Authorship
Contribution: M.B. and K.D. designed the study, collected data, and led the study; J.S.B., R.F.B., A.S., M.B., and K.D. wrote the manuscript; R.F.B. and M.B. performed statistical analysis; J.S.B., K.D., A.A.S., D.S.J, T.E.-G., and H.E.J. provided biological and clinical interpretation; M.P.-A., A.O., A.S., R.F.B., A.A.S., M.S., C.V., H.D., C.H.N., D.S.J., P.J., M.A.N., R.S., and R.J.S. contributed vital technology or analytical tools; M.K.S., N.M., F.D., B.W., C.P., M.K., D.J., G.J.M., U.B., H.G., A.B., M.v.D., and P.S. delivered clinical and expression data; all authors read and approved the final version of the manuscript before submission; and H.E.J. led the project before his death and therefore contributed in designing the study and drafting the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Hans Erik Johnsen died on 17 May 2018.
Correspondence: Martin Bøgsted, Department of Clinical Medicine, Aalborg University, Sdr. Skovvej 15, DK-9000 Aalborg, Denmark; e-mail: martin.boegsted@rn.dk.