Key Points
Novel CCND3 mutations were identified, and CDK4/6 inhibitors represent a promising therapeutic option for MLL-rearranged AML.
Coexisting mutations had a negative prognostic impact on pediatric MLL-MLLT3–rearranged AML patients.

Abstract
In acute myeloid leukemia (AML), MLL (KMT2A) rearrangements are among the most frequent chromosomal abnormalities; however, knowledge of the genetic landscape of MLL-rearranged AML is limited. In this study, we performed whole-exome sequencing (n = 9) and targeted sequencing (n = 56) of samples from pediatric MLL-rearranged AML patients enrolled in the Japanese Pediatric Leukemia/Lymphoma Study Group AML-05 study. Additionally, we analyzed 105 pediatric t(8;21) AML samples and 30 adult MLL-rearranged AML samples. RNA-sequencing data from 31 patients published in a previous study were also reanalyzed. As a result, we identified 115 mutations in pediatric MLL-rearranged AML patients (2.1 mutations/patient), with mutations in signaling pathway genes being the most frequently detected (60.7%). Mutations in genes associated with epigenetic regulation (21.4%), transcription factors (16.1%), and the cohesin complex (8.9%) were also commonly detected. Novel CCND3 mutations were identified in 5 pediatric MLL-rearranged AML patients (8.9%) and 2 adult MLL-rearranged AML patients (3.3%). Recurrent mutations of CCND1 (n = 3, 2.9%) and CCND2 (n = 8, 7.6%) were found in pediatric t(8;21) AML patients, whereas no CCND3 mutations were found, suggesting that D-type cyclins exhibit a subtype-specific mutation pattern in AML. Treatment of MLL-rearranged AML cell lines with CDK4/6 inhibitors (abemaciclib and palbociclib) blocked G1 to S phase cell-cycle progression and impaired proliferation. Pediatric MLL-MLLT3–rearranged AML patients with coexisting mutations (n = 16) had significantly reduced relapse-free survival and overall survival compared with those without coexisting mutations (n = 9) (P = .048 and .046, respectively). These data provide insights into the genetics of MLL-rearranged AML and suggest therapeutic strategies.
Introduction
Acute myeloid leukemia (AML) is a genetically and clinically heterogeneous disease characterized by expansion of undifferentiated myeloid precursor cells.1-3 Recurrent chromosomal and genetic abnormalities, including t(8;21), inv(16), t(15;17), and FLT3-internal tandem duplication (FLT3-ITD), are well-established markers for risk-stratified therapy in AML4,5 ; however, relapse remains common, and patients with relapsed AML have a poor prognosis.6 Recent genome-wide analyses revealed several recurrently mutated genes in AML, including DNMT3A, IDH1, IDH2, splicing factors, and cohesin complex genes7-9 ; however, few of these driver mutations have been developed as therapeutic targets to date. Moreover, because of the high level of genetic heterogeneity in AML, the clinical impact of driver mutations in each AML subtype remains unclear.
Rearrangements involving the MLL (mixed-lineage leukemia; official symbol, KMT2A) gene are among the most common chromosomal abnormalities in AML; these rearrangements are more common in pediatric AML (20%-24%), particularly infant AML (38%), than in adult AML (5%-10%),10-13 and >60 different MLL partner genes have been identified.14 Although MLL rearrangement has been regarded as a marker for poor prognosis in patients with AML, a recent study showed that the prognoses of patients with these changes vary considerably according to the MLL fusion partner. For example, pediatric AML patients with t(1;11)(q21;q23)/MLL-MLLT11 have favorable prognoses, whereas those with t(6;11)(q27;q23)/MLL-MLLT4(AF6), t(4;11)(q21;q23)/MLL-AFF1, t(10;11)(p12;q23)/MLL-MLLT10(AF10), and t(10;11)(p11.2;q23)/MLL-ABI1 have poor prognoses.15 The number of coexisting mutations is lower in MLL-rearranged AML than in other AML subtypes.2,13 RAS pathway mutations are well described; however, little is known about the prognostic significance of coexisting mutations in MLL-rearranged AML.16-18 A recent study of adult MLL-rearranged AML revealed novel somatic mutations in the transcription factor gene SPI1.18 Nevertheless, the genetic landscape of pediatric MLL-rearranged AML has yet to be fully elucidated, and further investigations are required to identify novel recurrent mutations and support improved therapeutic approaches in pediatric MLL-rearranged AML.
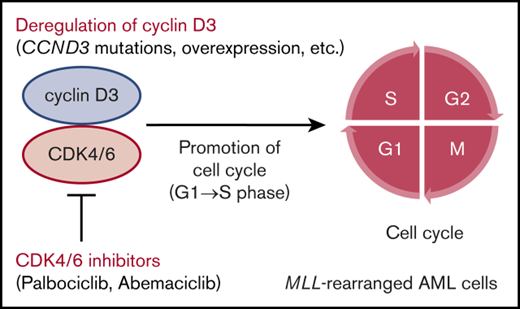
In this study, we identified recurrent mutations in CCND3, which encodes cyclin D3, in MLL-rearranged AML. Cyclins are cofactors that bind to cyclin-dependent kinases (CDKs), allowing cell-cycle progression. D-type cyclins form complexes with CDK4/6, which promote cell-cycle progression from G1 to S phase by phosphorylation of retinoblastoma protein (Rb).19,20 There are 3 D-type cyclins: cyclin D1 (encoded by CCND1), cyclin D2 (encoded by CCND2), and cyclin D3. Aberrant cell-cycle regulation mediated by deregulation of D-type cyclins is a major characteristic of cancer.21 In AML, recurrent mutations in CCND1 and CCND2 in t(8;21) AML were reported in previous studies22,23 ; however, recurrent CCND3 mutations have not been reported. A recent study showed that patients with deregulation of D-type cyclins may benefit from treatment with the CDK4/6 inhibitor24 ; therefore, we also examined the effectiveness of CDK4/6 inhibitors in MLL-rearranged AML cells.
Methods
Patients and study protocol
The AML-05 study is a Japanese nationwide multi-institutional study of children (age <18 years) with de novo AML conducted by the Japanese Pediatric Leukemia/Lymphoma Study Group (JPLSG) in which 485 patients were enrolled between 1 November 2006 and 31 December 2010. The trial was registered with the UMIN Clinical Trials Registry (#UMIN000000511; http://www.umin.ac.jp/ctr/index.htm), and details of the schedules and treatment regimens have been described previously.25 AML was diagnosed via a central review system. The study was conducted in accordance with the principles set down in the Declaration of Helsinki and approved by the ethics committees of all participating institutions. All patients, or their parents/guardians, provided written informed consent.
Whole-exome sequencing
Whole-exome sequencing was performed as previously reported.26 Whole-exome capture was accomplished by liquid phase hybridization of sonicated genomic DNA using a bait complementary RNA library (SureSelect Human ALL Exon V5 or V5 Inc RNA 5 kit; Agilent Technologies, Santa Clara, CA) following the manufacturer’s protocol. Massively parallel sequencing of the captured targets was performed using a HiSeq 2000/2500 sequencing system (Illumina, San Diego, CA) with the paired-end 126- to 133-bp read option, following the manufacturer’s instructions. Sequencing reads were aligned to hg19 using BWA-mem version 0.5.8 with default parameters. Details of the methods used to detect candidate somatic mutations using EBcell (Empirical Bayesian mutation Calling)27 are presented in supplemental Methods.
Targeted sequencing
In total, 338 gene targets were examined for gene mutations in the 56 MLL-rearranged AML pediatric cases using targeted capture sequencing and selected if they were (1) known driver genes in myeloid malignancies or other neoplasms, (2) associated with myeloid malignancies, (3) mutated as detected by whole-exome sequencing in this study, and (4) therapeutically targetable.
Sample preparation, sequencing, and data analyses were performed using the same approach as for whole-exome sequencing. Target enrichment was performed using a SureSelect custom kit (Agilent Technologies), which is designed to capture all coding exons of the above 338 genes and 1216 single-nucleotide polymorphisms for copy-number analysis. Similarly, CCND1, CCND2, and CCND3 were also captured and sequenced in samples from 105 pediatric cases with t(8;21)/RUNX1-RUNX1T1 AML and 30 adult patients with MLL-rearranged AML. Detailed methods for detecting candidate somatic mutations are presented in supplemental Methods.
Detection of copy-number alterations
Copy-number analysis was performed as previously reported28 using an in-house pipeline (Y. Shiozawa and S.O., manuscript in preparation) in which the total number of reads covering each bait region and the allele frequency of heterozygous single-nucleotide polymorphisms detected by targeted sequencing were used as input data.
Sanger sequencing and deep sequencing
First, polymerase chain reaction (PCR) was performed using a pair of primers targeting the PEST domain–encoding region of the CCND3 gene (forward, 5′-TGAGAACTAAAGAGCGATTCCTGG-3′; reverse, 5′-CTTTGTGAAGGGGGAACAGACG-3′). Reactions were performed in a volume of 20 μL containing 2 μL 10× PCR buffer for KOD plus polymerase, 2 μL 2′-deoxynucleoside 5′-triphosphate mix (2 mM), 1.2 µL MgSO4 (25 mM), 0.4 µL KOD plus polymerase (1 U/µL) (Toyobo, Osaka, Japan), 0.2 μL each primer (100 µM; Invitrogen, San Diego, CA), and 20 ng template DNA. Reactions were carried out in a Veriti 96-Well Thermal Cycler (Applied Biosystems, Foster City, CA) using a touchdown PCR protocol (1 cycle of 96°C for 2 minutes; 3 cycles of 96°C for 10 seconds, 64°C for 10 seconds, and 70°C for 30 seconds; 3 cycles of 96°C for 10 seconds, 61°C for 10 seconds, and 70°C for 30 seconds; 3 cycles of 96°C for 10 seconds, 58°C for 10 seconds, and 70°C for 30 seconds; 35 cycles of 96°C for 10 seconds, 57°C for 10 seconds, and 70°C for 30 seconds; and 1 cycle of 70°C for 5 minutes). PCR products were analyzed by agarose gel electrophoresis and purified using a FastGene Gel/PCR Extraction Kit (NIPPON Genetics, Tokyo, Japan) according to the manufacturer’s instructions. The sequences of purified PCR products were determined by direct sequencing using a forward primer (5′-CCAGACTTCCCCATGTGTTGG-3′) and a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) on a 3130xl Genetic Analyzer (Applied Biosystems). For deep sequencing, PCR amplicons were sonicated and prepared using a NEBNext Ultra DNA Library Prep Kit for Illumina (New England Biolabs). Sequencing was performed using a MiSeq platform with the 77-bp paired-end read option.
Compounds
Palbociclib (PD0332991) and abemaciclib (LY2835219) were obtained from AdooQ BioScience (Irvine, CA). Both compounds were dissolved in dimethyl sulfoxide (DMSO).
Cell culture
ML-2, MV4-11, and MOLM-13 cell lines were obtained from the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). THP-1 and NOMO-1 cell lines were obtained from the Japanese Collection of Research Bioresources Cell Bank (Ibaraki, Japan). All cell lines were cultured in RPMI 1640 medium containing 10% fetal bovine serum and 1% penicillin/streptomycin under 5% CO2 and 95% air at 37°C.
Cell proliferation assay
Cells (2 × 105/mL) were cultured in the presence of DMSO (control), palbociclib (500 nM), or abemaciclib (500 nM). Data are presented as the mean ± standard error of 3 independent experiments.
Cell-cycle analysis
Cells (2 × 105/mL) were treated with DMSO (control), palbociclib (500 nM), or abemaciclib (500 nM) for 24 hours. Then, cells were stained with propidium iodide and analyzed using a FACS Canto II flow cytometer (BD Biosciences, San Jose, CA).
Immunoblot analysis
Cells were washed with PBS and then lysed in RIPA buffer containing a protease inhibitor cocktail (Nakalai, Kyoto, Japan). After centrifugation, the protein content in supernatants was measured using a DC Protein Assay (Bio-Rad Laboratories, Hercules, CA). Whole-cell lysates containing equal amounts of total protein were separated on 10% sodium dodecyl sulfate polyacrylamide gels and then transferred to Immobilon-P transfer membranes (Merck, Darmstadt, Germany). Membranes were blocked with Blocking One reagent (Nakalai) for 1 h, followed by incubation overnight at 4°C with an anti-cyclin D3 antibody (1/1000; K0013-3; MBL, Nagoya, Japan) or an anti-GAPDH antibody (1/3000; sc-47724; Santa Cruz Biotechnology, Santa Cruz, CA). After washing thoroughly in Tris-buffered saline with Tween 20, membranes were incubated with horseradish peroxidase–conjugated whole anti-mouse immunoglobulin G (1/4000; NA931; GE Healthcare Bio-Sciences) for 1 hour at room temperature. Immunoreactive proteins were detected using a horseradish peroxidase Novex ECL Chemiluminescent Substrate Regent Kit (Invitrogen). Signals were captured, and the intensities of bands were quantified using a ChemiDoc XRS System (Bio-Rad Laboratories).
RNA interference
Small interfering RNA (siRNA) against human cyclin D3 transcript (sc-35136) and the nontargeting siRNA (sc-37007) were purchased from Santa Cruz Biotechnology. THP-1 cells (4 × 104/mL) were cultured in RPMI 1640 medium containing 10% fetal bovine serum without 1% penicillin/streptomycin at 37°C. After 1 day, THP-1 cells were transfected with siRNA/Lipofectamine RNAiMAX (Invitrogen) complexes diluted in Opti-MEM Reduced Serum Medium (Gibco) (final siRNA concentration, 50 nM) according to the manufacturer’s protocol. Two days after transfection, whole-cell lysates were prepared to determine knockdown efficiency by immunoblot analysis. Four days after transfection, THP-1 cells were collected for cell-cycle analysis by fluorescence-activated cell sorting.
Statistical analysis
Survival analyses were performed using the Kaplan-Meier method, and groups were compared using an unstratified log-rank test. Continuous variables were compared using the Student t test.
Results
Spectrum of mutations and copy-number alterations in pediatric MLL-rearranged AML
First, we analyzed paired AML tumor and germline samples from 9 pediatric MLL-rearranged AML patients enrolled in the JPLSG AML-05 study by whole-exome sequencing. Five of the patients were included in our previous study (supplemental Figure 1).29 In total, 52 mutations (mean, 5.8 mutations/patient) were identified (supplemental Table 1; supplemental Figure 2), including known mutational targets in AML, such as genes associated with signaling activation (FLT3 and BRAF), epigenetic regulation (SETD2 and BCORL1), and encoding transcription factors (WT1). Moreover, CCND3, which is a well-known driver gene in lymphomas,30 was mutated in 1 patient.
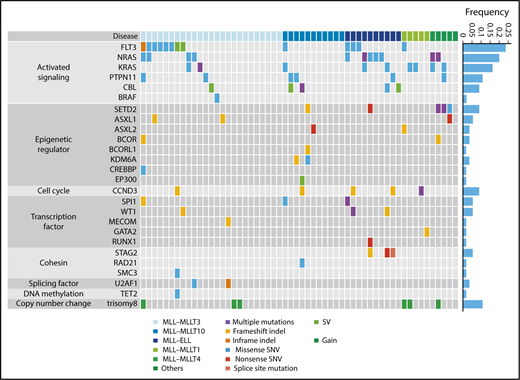
Next, we analyzed 56 samples from patients with pediatric MLL-rearranged AML enrolled in the JPLSG AML-05 study using targeted sequencing. Patient characteristics are presented in Table 1. The median age of patients was 3.0 years, which is consistent with data demonstrating that MLL-rearranged AML is frequent in infants.13 Over half of patients were classified in the M5a category (54%), followed by M4 (23%), M1 (7%), M5b (5%), M2 (2%), M4Eo (2%), and RAEB-T (2%). Among fusion types, MLL-MLLT3 was the most frequent (45%), followed by MLL-MLLT10 (20%), MLL-ELL (18%), MLL-MLLT1 (9%), and MLL-MLLT4 (AF6) (5%). We selected 338 genes, including CCND3, for targeted sequencing (supplemental Table 2). The driver or recurrent mutations detected by targeted sequencing are presented according to fusion type in Figure 1. Detailed results of targeted sequencing are provided in supplemental Table 3. We identified 115 mutations (mean, 2.1 mutations/patient), with the following mutations in signaling pathway genes being the most frequently detected (60.7%): FLT3 (n = 13, 23.2%), NRAS (n = 11, 19.6%), KRAS (n = 9, 16.1%), PTPN11 (n = 6, 10.7%), CBL (n = 5, 8.9%), and BRAF (n = 1, 1.8%). Mutations in genes associated with epigenetic regulation (SETD2, ASXL1, ASXL2, BCOR, BCORL1, KDM6A, CREBBP, and EP300) (21.4%), transcription factors (SPI1, WT1, MECOM, GATA2, and RUNX1) (16.1%), and the cohesin complex (STAG2, RAD21, and SMC3) (8.9%) were also commonly detected. Interestingly, all MLL-ELL patients had at least 1 mutation of a signaling pathway gene, and all 3 STAG2 mutations were restricted to MLL-ELL patients, suggesting that the distribution of coexisting mutations differs according to MLL fusion partner.
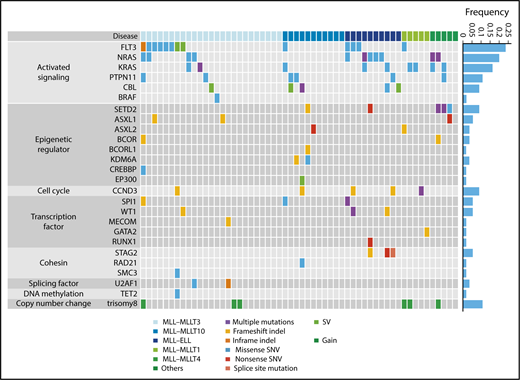
Mutational landscape of pediatric MLL-rearranged AML. Driver or recurrent mutations and copy-number alterations observed in pediatric MLL-rearranged AML (n = 56). The frequencies of mutated genes are shown on the right. SNV, single-nucleotide variant; SV, structural variant.
Mutational landscape of pediatric MLL-rearranged AML. Driver or recurrent mutations and copy-number alterations observed in pediatric MLL-rearranged AML (n = 56). The frequencies of mutated genes are shown on the right. SNV, single-nucleotide variant; SV, structural variant.
Copy-number alterations were also infrequent in MLL-rearranged AML, with trisomy 8 being the most frequently detected (identified in 6 cases; Figure 1; supplemental Figure 3), which is consistent with previous reports showing that trisomy 8 is among the most common cytogenetic aberrations in pediatric AML.31,32
Recurrent CCND3 mutations in pediatric and adult MLL-rearranged AML
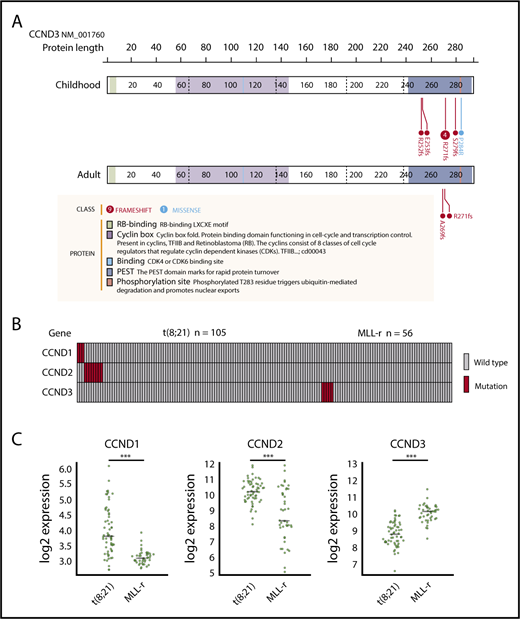
We identified 8 mutations in CCND3, which encodes cyclin D3, in 5 pediatric MLL-rearranged AML patients (8.9%). Detailed information regarding patients with CCND3 mutations is presented in Table 2. All mutations were clustered in the PEST domain (Figure 2A), which is found in short-lived proteins and plays a role in their degradation.33 Four of the 8 mutations were R271fs, which is a known hot-spot mutation in lymphoid malignancies.30,34-38
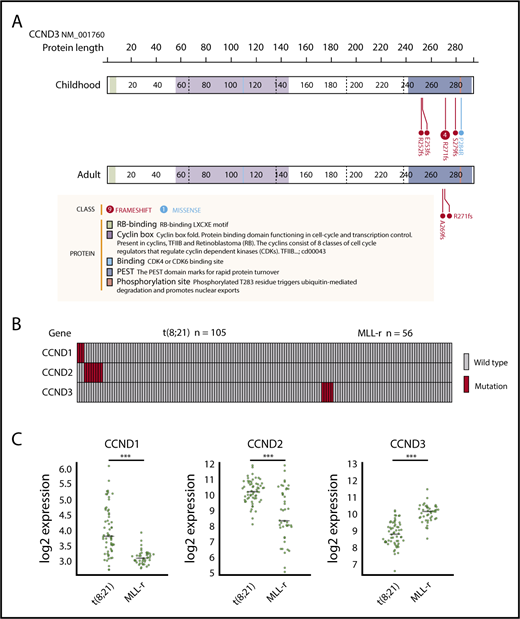
CCND3 mutations in MLL-rearranged AML and disease subtype–specific pattern of CCND1, CCND2, and CCND3 mutations and expression. (A) Domain structure and location of CCND3 mutations in pediatric and adult MLL-rearranged AML. Lollipop plots were generated using ProteinPaint (https://pecan.stjude.org/proteinpaint/). (B) Mutations of CCND1, CCND2, and CCND3 in pediatric AML patients with t(8;21) and MLL rearrangements (MLL-r). (C) Messenger RNA expression levels of CCND1, CCND2, and CCND3 in t(8;21) AML (n = 60) and MLL-rearranged AML (n = 43). ***P < .001.
CCND3 mutations in MLL-rearranged AML and disease subtype–specific pattern of CCND1, CCND2, and CCND3 mutations and expression. (A) Domain structure and location of CCND3 mutations in pediatric and adult MLL-rearranged AML. Lollipop plots were generated using ProteinPaint (https://pecan.stjude.org/proteinpaint/). (B) Mutations of CCND1, CCND2, and CCND3 in pediatric AML patients with t(8;21) and MLL rearrangements (MLL-r). (C) Messenger RNA expression levels of CCND1, CCND2, and CCND3 in t(8;21) AML (n = 60) and MLL-rearranged AML (n = 43). ***P < .001.
To determine whether CCND3 mutations were also present in adult MLL-rearranged AML, we analyzed 30 adult MLL-rearranged AML samples for CCND3 mutations by targeted sequencing (patient information provided in supplemental Table 4). RNA-sequencing data from 31 patients published in a previous study were also reanalyzed.18 CCND3 mutations affecting the PEST domain were detected in 2 out of 61 samples (3.3%) (Figure 2A). In total, 4 out of 7 patients (57.1%) with CCND3 mutations were detected among those with MLL-ELL–positive AML. All CCND3 mutations identified in our cohort were confirmed by Sanger sequencing or deep sequencing (supplemental Figure 4). Mutations of the PEST domain of CCND3 elevate the stability of cyclin D3 protein and promote cell proliferation30 ; therefore, these results suggest that the stabilization of cyclin D3 is important in both pediatric and adult MLL-rearranged AML.
Mutations of D-type cyclins exhibit a disease subtype–specific pattern in AML
Mutations of the other D-type cyclins (CCND1 and CCND2) have been reported in t(8;21) AML22,23 ; therefore, we also searched for mutations in CCND1, CCND2, and CCND3 by targeted sequencing of samples from pediatric AML patients with t(8;21)/RUNX1-RUNX1T1 (n = 105). CCND1 mutations (n = 3; 2.9%) and CCND2 mutations (n = 8; 7.6%) were detected in t(8;21) patients; however, no mutations of CCND3 were found in this group (Figure 2B; supplemental Figure 5). By contrast, there were no mutations of CCND1 or CCND2 in MLL-rearranged AML (n = 56), suggesting that mutations of D-type cyclins exhibit a subtype-specific pattern in AML.
Furthermore, we compared the messenger RNA expression levels of CCND1, CCND2, and CCND3 between t(8;21) AML (n = 60) and MLL-rearranged AML (n = 43) (in dataset GSE13159, generated as part of the MILE [Microarray Innovations in LEukemia] study program) using BloodSpot.39 CCND1 and CCND2 expression was significantly higher in t(8;21) AML than MLL-rearranged AML (both P < .001), while CCND3 expression was significantly higher in MLL-rearranged AML than t(8;21) AML (P < .001) (Figure 2C). These data support the hypothesis that deregulation of D-type cyclins exhibits a subtype-specific pattern in AML.
CDK4/6 inhibitors are a promising treatment of MLL-rearranged AML
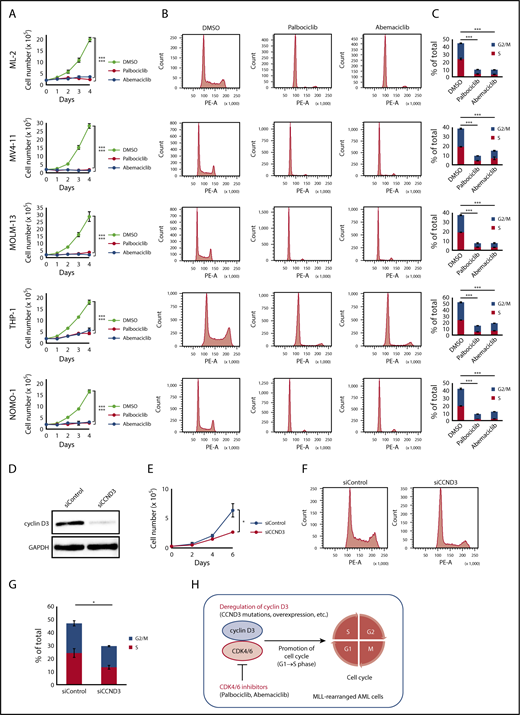
A recent study demonstrated that cancers with CCND3 amplification may benefit from treatment with the CDK4/6 inhibitor abemaciclib24 ; therefore, we examined whether CDK4/6 inhibitors could be novel therapeutic agents for MLL-rearranged AML. The effects of major CDK4/6 inhibitors (abemaciclib and palbociclib) on the proliferation and cell-cycle progression of MLL-rearranged AML cell lines (ML-2 [MLL-MLLT4 positive], MV4-11 [MLL-AFF1 positive], MOLM-13 [MLL-MLLT3 positive], THP-1 [MLL-MLLT3 positive], and NOMO-1 [MLL-MLLT3 positive]) were analyzed. The CDK4/6 inhibitor concentration employed was 500 nM, based on previously published data.40 Among these cell lines, a CCND3 mutation (R271fs) was detected in ML-2 cells by Sanger sequencing (supplemental Figure 4). CCND3/cyclin D3 was highly expressed in MLL-rearranged compared with t(8;21) AML cell lines (Kasumi-1 and SKNO-1) (supplemental Figure 6). MLL-rearranged AML cell lines exhibited impaired proliferation after treatment with CDK4/6 inhibitors (Figure 3A). Furthermore, treatment of these cell lines with CDK4/6 inhibitors resulted in detection of lower frequencies of S/G2/M phase cells by flow cytometry, suggesting that cells were arrested in G1 phase via CDK4/6 inhibition (Figure 3B-C).
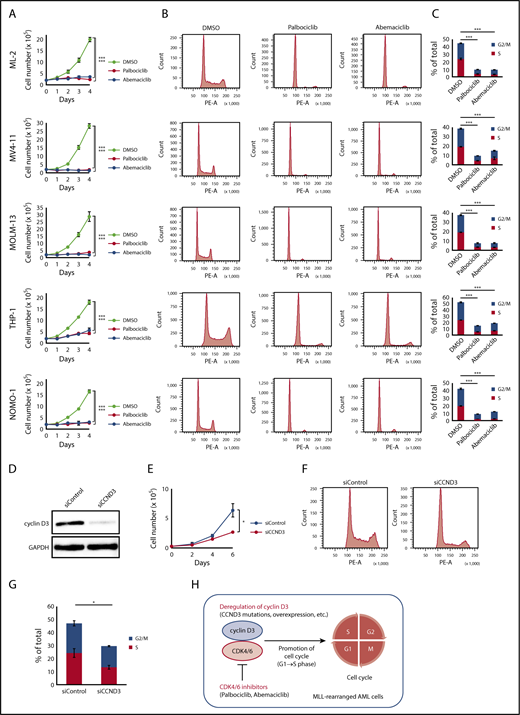
Effects of CDK4/6 inhibitors in MLL-rearranged AML cell lines. (A) Cell proliferation assay. ML-2, MV4-11, MOLM-13, THP-1, and NOMO-1 cells were cultured in the presence of DMSO, palbociclib (500 nM), or abemaciclib (500 nM). (B) Cell-cycle analysis. Flow cytometric analysis of cell lines treated with DMSO, palbociclib (500 nM), or abemaciclib (500 nM) for 24 hours and stained with propidium iodide. (C) Comparison of percentages of G2/M and S phase cells. (D) Immunoblot analysis revealed significantly lower expression of cyclin D3 proteins in THP-1 cells transfected with siRNA against human cyclin D3 transcript (left column: siCCND3) than in THP-1 cells transfected with non-targeting siRNA (right column: siControl). (E-G) THP-1 cells transfected with siCCND3 showed significant impairment in cell proliferation (E) and cell-cycle progression from G1 to S phase (F-G) compared with those transfected with nontargeting siRNA. (H) Schematic figure showing that the cyclin D3-CDK4/6 complex promotes cell-cycle progression and CDK4/6 inhibitors are promising therapeutic agents in MLL-rearranged AML cells. Data are presented as the mean ± standard error of 3 independent experiments. ***P < .001, *P < .05.
Effects of CDK4/6 inhibitors in MLL-rearranged AML cell lines. (A) Cell proliferation assay. ML-2, MV4-11, MOLM-13, THP-1, and NOMO-1 cells were cultured in the presence of DMSO, palbociclib (500 nM), or abemaciclib (500 nM). (B) Cell-cycle analysis. Flow cytometric analysis of cell lines treated with DMSO, palbociclib (500 nM), or abemaciclib (500 nM) for 24 hours and stained with propidium iodide. (C) Comparison of percentages of G2/M and S phase cells. (D) Immunoblot analysis revealed significantly lower expression of cyclin D3 proteins in THP-1 cells transfected with siRNA against human cyclin D3 transcript (left column: siCCND3) than in THP-1 cells transfected with non-targeting siRNA (right column: siControl). (E-G) THP-1 cells transfected with siCCND3 showed significant impairment in cell proliferation (E) and cell-cycle progression from G1 to S phase (F-G) compared with those transfected with nontargeting siRNA. (H) Schematic figure showing that the cyclin D3-CDK4/6 complex promotes cell-cycle progression and CDK4/6 inhibitors are promising therapeutic agents in MLL-rearranged AML cells. Data are presented as the mean ± standard error of 3 independent experiments. ***P < .001, *P < .05.
Next, we confirmed the role of cyclin D3 in MLL-rearranged AML cells. Among the 5 MLL-rearranged AML cell lines, THP-1 was selected because it could be knocked down efficiently by siRNA (Figure 3D). THP-1 cells transfected with siRNA against the human cyclin D3 transcript showed significant reductions in cell proliferation and cell cycling from G1 to S phase compared with THP-1 cells transfected with nontargeting siRNA (Figure 3E-G). Taken together, the results suggest that the cyclin D3-CDK4/6 complex plays an important role in cell-cycle progression in MLL-rearranged AML and that cyclin D3 and CDK4/6 are essential for the mechanism (Figure 3H).
Coexisting mutations are associated with poor prognosis in MLL-MLLT3–rearranged AML
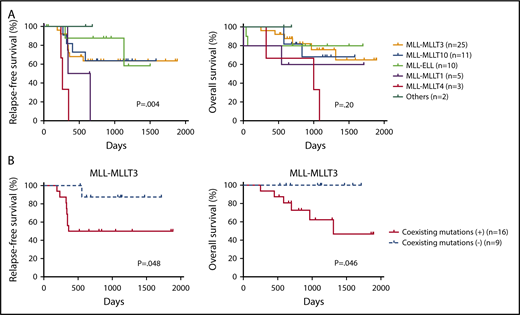
Finally, we examined the prognostic significance of fusion partners and coexisting mutations in pediatric MLL-rearranged AML. Consistent with a previous report, patients with MLL-MLLT4 exhibited poor outcomes (Figure 4A).15 The relapse-free survival (RFS) of patients with MLL-MLLT4 was significantly shorter than that of those with MLL-MLLT3 (P = .004), MLL-MLLT10 (P = .001), or MLL-ELL (P = .002). Interestingly, patients with MLL-MLLT1 also had poor outcomes, and the RFS of patients with this rearrangement was significantly shorter than that of patients with MLL-ELL (P = .03). P values for comparisons between patients with each fusion subtype are provided in supplemental Table 5.
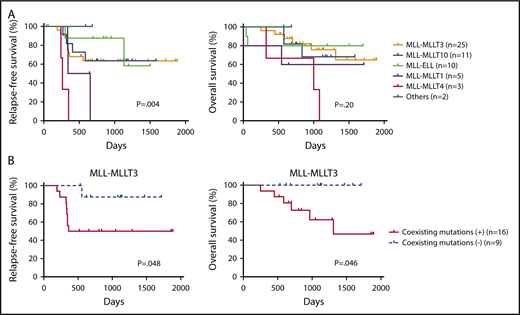
Prognostic significance of fusion partners in pediatric MLL-rearranged AML and coexisting mutations in pediatric MLL-MLLT3–rearranged AML. (A) Comparison of patient RFS and OS according to MLL fusion partner. (B) Comparison of RFS and OS for patients with pediatric MLL-MLLT3–rearranged AML with and without coexisting mutations. Survival estimates were compared using the log-rank test.
Prognostic significance of fusion partners in pediatric MLL-rearranged AML and coexisting mutations in pediatric MLL-MLLT3–rearranged AML. (A) Comparison of patient RFS and OS according to MLL fusion partner. (B) Comparison of RFS and OS for patients with pediatric MLL-MLLT3–rearranged AML with and without coexisting mutations. Survival estimates were compared using the log-rank test.
We also evaluated the prognostic significance of coexisting mutations identified in MLL-MLLT3–rearranged AML (n = 25), because this rearrangement was the most common in the cohort. Patients with coexisting mutations listed in Figure 1 (n = 16) had significantly reduced RFS and overall survival (OS) compared with those without coexisting mutations (n = 9) (P = .048 and .046, respectively) (Figure 4B). Investigation of the prognostic impact of mutated pathways indicated that the RFS and OS of patients with activated signaling mutations (FLT3, NRAS, KRAS, PTPN11, CBL, and BRAF) tended to be shorter than those of patients without such mutations; however, the difference was not statistically significant (supplemental Figure 7). We also analyzed the prognostic significance of mutations of FLT3, CCND3, and RAS pathway (NRAS, KRAS, PTPN11, and CBL), epigenetic regulator (SETD2, ASXL1, ASXL2, BCOR, BCORL1, KDM6A, CREBBP, and EP300), transcription factor (SPI1, WT1, MECOM, GATA2, and RUNX1), and cohesin complex (STAG2, RAD21, and SMC3) genes (supplemental Figure 8). No significant differences in outcomes were associated with these mutation types. We also analyzed the prognostic significance of coexisting mutations in other subgroups (MLL-MLLT10, MLL-ELL, MLL-MLLT1, and MLL-MLLT4); however, there were no significant differences (data not shown). These analyses were limited by the small cohort size, therefore, further studies with larger cohorts should be conducted.
Discussion
In this study, we performed whole-exome sequencing of samples from 9 pediatric patients with MLL-rearranged AML. The number of mutations was smaller (mean, 5.8 mutations/patient) compared with our previous findings from pediatric t(8;21) AML samples (mean, 7.8 mutations/patient) (n = 5)29 and data from a cohort of pediatric core binding factor AML samples (mean, 7.4 mutations/sample) (n = 87).22 This suggests that MLL fusion proteins require fewer cooperating mutations than t(8;21) or inv(16).
We also performed targeted sequencing of 338 genes in 56 pediatric MLL-rearranged AML. Among the detected mutations, alterations of signaling pathway genes were the most frequently detected (34/56 patients; 60.7%), which is consistent with previous studies.16,17 A BRAF V600E mutation was detected in 1 patient. This mutation is found in some types of cancer, including hairy cell leukemia, and is a promising therapeutic target41 ; therefore, screening for the BRAF V600E mutation should be considered in MLL-rearranged AML. Patients with MLL-MLLT3-rearranged AML and coexisting mutations had significantly worse RFS and OS. MLL-MLLT3–rearranged AML is categorized as an intermediate-risk group15 ; therefore, screening for coexisting mutations may enable improved risk assessment for these patients. Previous studies failed to determine the prognostic significance of coexisting mutations in MLL-rearranged AML, which may be because of the limited numbers of cases of each fusion partner included in the analyses.16,17 Further studies including larger numbers of patients are required.
We also observed recurrent mutations of epigenetic regulator genes (SETD2, ASXL1, ASXL2, BCOR, BCORL1, KDM6A, CREBBP, and EP300) in 12 patients (21.4%), suggesting that aberrant epigenetic regulation is strongly involved in MLL-rearranged AML pathogenesis. The result is not surprising, given its role as a histone methyltransferase. CREBBP and EP300 encode highly related acetyltransferases that act as transcriptional coactivators in multiple signaling pathways.42 Recurrent mutations of CREBBP and EP300 are reported in lymphomas and acute lymphoblastic leukemia (ALL); however, few studies have identified these mutations in AML, except for rare translocations involving CREBBP and EP300.35,43-46 Together with the recurrent CCND3 mutations detected in this study, these data indicate that MLL-rearranged AML may share some genetic features with lymphomas and ALL.
Mutations in cohesin complex genes (STAG2, RAD21, and SMC3) were detected in 5 patients (8.9%). Cohesin is a multimeric protein complex composed of 4 subunits: STAG2, RAD21, SMC1A, and SMC3.47 The frequency of mutations in cohesin complex genes is very high in Down syndrome–related acute megakaryoblastic leukemia.48 In de novo AML, the frequency of cohesin complex mutations is 10% to 12%,9,49 which is slightly higher than our findings for MLL-rearranged AML; however, STAG2 mutations were detected in 3 patients with MLL-ELL, equating to a frequency of 30%. Furthermore, in a previous study of adult MLL-rearranged AML, both STAG2 mutations identified were also restricted to patients with MLL-ELL.18 Hence, although the underlying mechanism is unclear, STAG2 mutations may have an important role in the pathogenesis of MLL-ELL–rearranged AML.
We identified recurrent CCND3 mutations in pediatric and adult MLL-rearranged AML. The frequency of CCND3 mutations in adult patients (2/61; 3.3%) was lower than that in pediatric patients (5/56; 8.9%), which may be attributable to the different distribution of translocation partners between these groups (Table 1; supplemental Table 4). CCND3 mutations are well described in lymphoid malignancies, including Burkitt lymphoma, diffuse large B-cell lymphoma, follicular lymphoma, hairy cell leukemia variant (HCL-V), splenic diffuse red pulp lymphoma, and T-lineage ALL.30,34-38 Nevertheless, there are few reports of CCND3 mutations in AML other than 1 description of a case with normal karyotype AML and CCND3 deletion.50 All CCND3 mutations detected in this study were clustered in the region encoding the PEST domain, similar to observations in lymphoid malignancies. CCND3 mutations affecting the PEST domain cause stabilization of the cyclin D3 protein, resulting in cell-cycle deregulation30 ; therefore, this mechanism may be associated with the pathogenesis of MLL-rearranged AML, as well as that of lymphoid malignancies. Mutation and overexpression of CCND1, CCND2, and CCND3 exhibited an AML subtype–specific pattern. In a previous study, selective use of D-type cyclins was revealed in lymphoid malignancies by analysis of numerous cell lines51 ; however, little is known about this phenomenon in AML. Our data suggest that particular pairs of deregulated D-type cyclins and chromosomal abnormalities may be associated with AML pathogenesis.
MLL-rearranged AML cell lines showed blocked G1 to S phase cell-cycle progression and impaired proliferation after treatment with CDK4/6 inhibitors (palbociclib and abemaciclib) (Figure 3). These inhibitors are currently in clinical trials for treatment of multiple cancers, including breast cancer and mantle cell lymphoma, and some reports have already demonstrated that their use is linked to improved survival rates.52-54 CCND3 amplification was recently identified as a promising marker for predicting responses to CDK4/6 inhibitors.24,55 In this study, CDK4/6 inhibitors were effective in inhibiting the growth of not only a CCND3-mutated cell line (ML-2) but also that of cell lines lacking CCND3 mutations (MV4-11, MOLM-13, THP-1, and NOMO-1). The MLL-rearrangement cell lines without CCND3 mutations expressed abundant cyclin D3 protein (supplemental Figure 6); therefore, CDK4/6 inhibitors may be effective for the treatment of malignancies with these characteristics. According to previous studies on other D-type cyclins (CCND1 and CCND2),24,56 increased CCND3 expression in cell lines without CCND3 mutations may be caused by mutations or posttranscriptional modifications in 3′ untranslated regions of CCND3. According to the response profiles of abemaciclib across 560 reported cancer cell lines, ∼20% of AML cells were sensitive; however, the subtype of AML exhibiting sensitivity was not discussed.24 This study revealed recurrent CCND3 mutations and high CCND3 expression in MLL-rearranged AML patients; therefore, CDK4/6 inhibitors represent a promising therapeutic option, particularly for this patient subgroup. These data provide further insights into the genetic basis of, and potential therapeutic strategies for, MLL-rearranged AML.
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported by a Grant-in-Aid from the Agency for Medical Research and Development (Project for Development of Innovative Research on Cancer Therapeutics [P-DIRECT], Project for Cancer Research And Therapeutic Evolution [P-CREATE]), the Japan Society for the Promotion of Science (KAKENHI grants 16K19193, 26221308, and 15H05909), a research grant from the Japanese Society of Hematology, and Kurozumi Medical Foundation. Supercomputing resources were provided by the Human Genome Center, Institute of Medical Science, The University of Tokyo.
Authorship
Contribution: H. Matsuo analyzed clinical data and performed the functional analysis; K.Y. analyzed the sequencing data; H. Matsuo and K.Y. wrote the manuscript; Y. Shiozawa, Y. Shiraishi, K.C., H.T., A.O., and S. Miyano developed sequence data processing pipelines; H. Matsuo, K.Y., K.F., H.U., and H. Mano performed sequencing; Y. Noguchi helped with statistical analyses; K.N., S.T., and M.N. helped with functional analyses; H. Matsuo, Y. Nannya, J.T., N.S., G.Y., H.H., Y.O., N.H., T.I., K.U., K.I., S. Miyawaki, H.I., Y.M., M.K., H.Y., N.K., D.T., T.T., A.T., Y.H., and S.A. collected clinical samples; and H. Mano, Y.K., S.O., and S.A. supervised the project.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Souichi Adachi, Department of Human Health Sciences, Graduate School of Medicine, Kyoto University, 53 Kawahara-cho, Syogoin, Sakyoku, Kyoto 606-8507, Japan; e-mail: adachiso@kuhp.kyoto-u.ac.jp.