Key Points
In patients with the genetically heterogeneous group of intermediate OP, HSCT offers the potential for cure and improved quality of life.

Abstract
Osteopetrosis (OP) is a rare disease caused by defective osteoclast differentiation or function. Hematopoietic stem cell transplantation (HSCT) is the only curative treatment available in the infantile “malignant” form of OP. Improved clinical and genetic diagnosis of OP has seen the emergence of a cohort of patients with less severe and heterogeneous clinical presentations. This intermediate form of OP does not call for urgent intervention, but patients accumulate debilitating skeletal complications over years and decades, which are severe enough to require curative treatment and may also require intermittent transfusion of blood products. Here we present data from 7 patients with intermediate OP caused by mutations in TCIRG1 (n = 2), CLCN7 (n = 2), RANK (n = 1), SNX10 (n = 1), and CA2 (n = 1), who were transplanted between the ages of 5 to 30 years (mean, 15; median, 12). Donors were matched siblings or family (n = 4), matched unrelated (n = 2), or HLA haploidentical family donors (n = 1). Conditioning was fludarabine and treosulfan based. All 6 patients transplanted from matched donors are currently alive with a follow-up period between 1 and 8 years at time of publication (median, 4 years) and have demonstrated a significant improvement in symptoms and quality of life. Patients with intermediate OP should be considered for HSCT.
Introduction
Osteopetrosis is a rare genetic disease characterized by defective osteoclast activity that leads to decreased bone resorption and a significant increase in bone mass. Defective osteoclast activity can be caused by mutations in genes affecting osteoclast development (eg, RANK, RANKL), and function (eg, TCIRG1, SNX10, CLCN7, OSTM1, CA2, PLEKHM1). Biallelic mutations in these genes account for >90% of all cases of malignant infantile osteopetrosis (MIOP),1-3 whereas autosomal dominant osteopetrosis is caused by monoallelic mutations in the CLCN7 gene. Although MIOP is early in onset and universally fatal without hematopoietic stem cell transplantation (HSCT), autosomal dominant osteopetrosis presents in later life with milder disease manifestations.
Patients with MIOP typically present with severe failure to thrive, macrocephaly, and visual impairment in infancy. Skull changes may lead to hydrocephalus, choanal stenosis, and compression of the optic nerve, resulting in blindness. Additional clinical symptoms and signs include micrognathia, small thorax, pathological fractures, hepatosplenomegaly, hypocalcemia, and severe dental defects. Bone marrow failure, as a result of bony encroachment into the hematopoietic niche, leads to thrombocytopenia and anemia.1-4
When studying the clinical and laboratory variability of osteopetrosis patients, a third group of patients emerges that can be neither classified as MIOP nor the benign dominant form of osteopetrosis. This intermediate form of osteopetrosis presents in children <10 years of age who are affected by recurrent pathological fractures and abnormal bone morphology, but tend not to develop bone marrow failure and extramedullary hematopoiesis. Children frequently experience severe debilitating morbidity requiring frequent hospitalizations, with considerable effect on their development, usually by the end of their first decade of life.5
The only curative option for MIOP is allogeneic HSCT. Successful HSCT leads to engraftment of macrophage-derived osteoclasts of donor origin, resulting in remodeling of bone structure and establishment of normal hematopoiesis.6 Different groups6-8 have previously published experience with HSCT in MIOP; however, the treatment strategy for patients suffering from intermediate osteopetrosis (IMO) has not been defined so far and data regarding HSCT are lacking. Here we present combined data from 2 centers with long-standing experience in treating patients with MIOP, and describe the clinical, genetic, and HSCT outcomes in 7 patients diagnosed with IMO.
Patients and methods
Three patients at the Hadassah-Hebrew University Medical Centre (Jerusalem, Israel) and 4 patients at the Ulm University Medical Centre (Ulm, Germany) with IMO were included between 2008 and 2016. Ethical review boards of both centers approved the study, and patients were treated after they or their parents provided written informed consent according to the Declaration of Helsinki.
Details of patient clinical status before HSCT are summarized in Table 1 and the supplemental Data. We report on 4 male and 3 female patients from 6 consanguineous families of Arab, Tajik, Dagestanian, and European origin. All patients demonstrated homozygous inheritance of defects in genes known to cause osteopetrosis: TCIRG1 (n = 2), CLCN7 (n = 2), CA2 (n = 1), SNX10 (n = 1), and RANK (n = 1). All patients developed features of osteopetrosis in the first year of life. The patients treated at Hadassah Medical Center (P1, P2, P7) were not formally diagnosed until being transferred to the tertiary center where whole exome sequencing confirmed the clinical suspicion of osteopetrosis; for P2, this took 8 years and for P7, it took 27 years. P1 is part of a large kindred with many cases of SNX10, all of whom presented with classic features of MIOP; thus, his parents recognized the features of the disease early in life but felt they were milder than in P1’s relatives, and he did not receive a genetic diagnosis until the SNX10 gene was described in 2012 (the patient was 11 years old at the time).9 In P3, blindness was diagnosed at birth; she experienced several pathological fractures and osteomyelitis of the jaw since infancy, but did not receive a genetic diagnosis until the age 11, when RANK mutations as the cause of MIOP were described in the literature.10 P4 accumulated pathological fractures in the first years of life, when a homozygous mutation in TCIRG1 was detected. Because of the absences of visual and hematological impairments, he was not considered for HSCT at the local hospital until the age of 29 years, when pain in the bones worsened and transfusion-dependent anemia occurred; he was transferred to the transplant center in Ulm. P5 and P6 are siblings of Arabic origin who showed impairments of vision but no transfusion dependency in infancy. They were transferred to the transplant center in Ulm at the age of 7 and 5 years, respectively, because of worsening of vision and bone marrow function. Six of 7 patients showed facial and dental abnormalities, hematological impairment, short stature (height <10th percentile), pathological fractures, and various degree of visual impairment. Three patients (P2, P3, P4) had developed bacterial infectious osteomyelitis and 2 (P1, P3) required tracheostomy to resolve respiratory impairment caused by choanal stenosis and jaw deformities. P1 suffered from hydrocephalus, necessitating ventriculoperitoneal shunt insertion. Only 1 patient had a complete loss of vision (P3, RANK mutation) and only the oldest patient (P4, 30 years, TCIRG1 mutation) received regular red blood cell transfusions at the time of HSCT (P2, P3, P6, and P7 required transfusions intermittently).
Indication for HSCT was made individually based on disease burden. All but 1 patient (P2) suffered from pathological fractures and imminent blindness, and 3 patients (P2, P3, P4) suffered from chronic destructive infectious osteomyelitis of the maxilla or mandible (computed tomography reconstruction of P3 in Figure 1A; P4 in supplemental Figure 1). Age at HSCT was between 5 and 30 years (mean, 15; median 12, years).
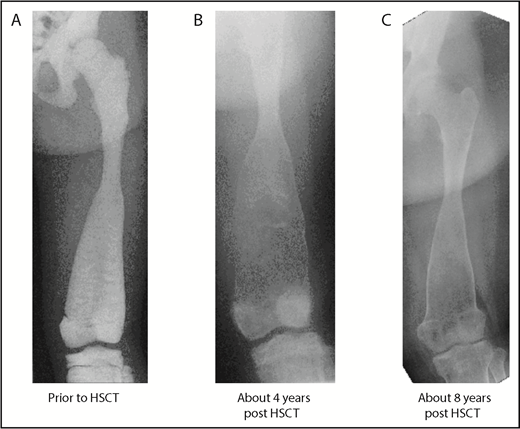
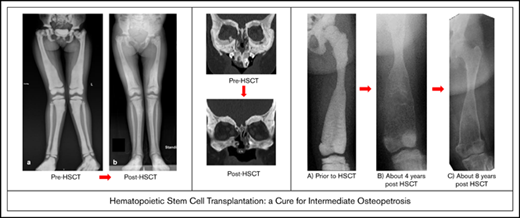
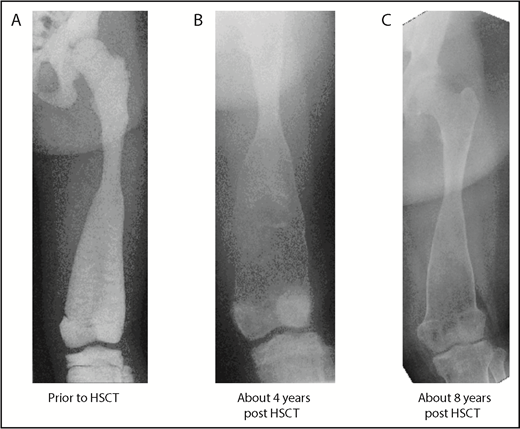
Baseline and follow-up plain radiography of the left lower extremity (femur) of patient 3. (A) Before HSCT, highly increased bone density with significantly diminished radiograph transparency, “ballooning,” especially of the distal femoral diaphysis, and meta/epiphysis. Transversal intraosseous dense horizontal lines are seen. (B) About 4 years after HSCT regression/normalization of bone density, however, decreased cortical lining and even increased osseous “ballooning” is seen. (C) About 8 years after HSCT, remodeling of bone size and complete normalization of cortical thickness is seen.
Baseline and follow-up plain radiography of the left lower extremity (femur) of patient 3. (A) Before HSCT, highly increased bone density with significantly diminished radiograph transparency, “ballooning,” especially of the distal femoral diaphysis, and meta/epiphysis. Transversal intraosseous dense horizontal lines are seen. (B) About 4 years after HSCT regression/normalization of bone density, however, decreased cortical lining and even increased osseous “ballooning” is seen. (C) About 8 years after HSCT, remodeling of bone size and complete normalization of cortical thickness is seen.
Characteristics of the HSCT procedures are summarized in Table 2. Donors were either matched sibling (n = 1), HLA-identical family members in consanguineous families (n = 3), matched unrelated donors (MUDs; n = 2; 9/10 or 10/10 HLA-compatible, respectively), or an HLA haploidentical family donor (n = 1). The graft consisted of unmanipulated bone marrow (n = 4) or peripheral blood stem cells (PBSC; n = 2) for HLA-matched grafts; the HLA haploidentical graft consisted of CD34+ selected PBSC. A combination of treosulfan, fludarabine, and thiotepa was used for conditioning in 6 patients transplanted with a T cell–replete graft; busulfan was used instead of treosulfan in the T cell–depleted HLA haploidentical setting (P6; Table 2) Serotherapy was given in all but 2 patients with antithymocyte globulin (ATG, n = 3) and alemtuzumab (Campath, n = 2). Graft-versus-host disease (GVHD) prophylaxis consisted of a combination of calcineurin inhibitors and mycophenolate mofetil (MMF) in all but the haploidentical, T cell–depleted case. In P7, who was transplanted from a 9/10 MUD, GVHD prophylaxis consisted of cyclosporine A (CSA), MMF, and cyclophosphamide post-HSCT. For prophylaxis of hepatic veno-occlusive disease (VOD)/sinusoidal obstruction syndrome (SOS), defibrotide was used in 4 patients (P3-P6).
Results
Details of the posttransplant course are depicted in Table 2. Six of 7 patients are alive. Median follow-up of the 6 surviving patients is 4 years (range, 1-8 years). All but 1 patient showed primary and sustained engraftment. Neutrophil engraftment (>0.5 × 109/L for 3 consecutive days) was reached 28 to 39 days post-HSCT (mean, 24; median, 25), whereas platelet engraftment (>20 × 109/L for 3 consecutive days) was reached 28 to 39 days after HSCT (mean and median, 33). All surviving patients demonstrate 100% donor chimerism at most recent follow-up. The only patient transplanted with a T cell–depleted haploidentical graft (P6) rejected her first graft and was retransplanted from the same donor after an immunosuppressive treatment using cyclophosphamide and the T-cell antibody Okt-3. After a second rejection, she was treated with fludarabine and antithymocyte globulin and transplanted a third time from another haploidentical donor, but died before engraftment from cytomegalovirus (CMV) disease and pulmonary hypertension.
One patient developed acute GVHD grade 2-3 and subsequently limited chronic GVHD (P3), which was successfully treated. Infectious complications were common, with bacterial sepsis or bacteremia in 5 patients, CMV reactivation in 3, and Epstein Barr virus–related disease in 1 patient. Of note, all 4 patients who were older than 10 years at the time of HSCT developed hypercalcemia during engraftment. This was treated with denosumab in 2 cases when conservative treatment with diuretics failed.11
Pathological fractures are reported for 2 patients merely in the first year of follow-up. All surviving patients showed a significant improvement in their clinical condition and quality of life; in particular, their hematological abnormalities improved (no further need for transfusion of blood or platelets). They suffered no further deterioration in their vision or hearing. Bone density and bone deformation improved over months and years in all patients (Figure 1; supplemental Figure 3), and no further pathological fracture occurred from 1 year after transplantation. Osteomyelitis of the jaw in P2, P3, and P4 resolved (reconstruction of P3 in Figure 1; P3 in supplemental Figure 2). Two patients dependent on a wheelchair (P3, P4) are walking again without assistance (supplemental Video). All patients were able to attend school or to go to work.
Discussion
In comparison with patients with MIOP, the indication for HSCT in patients with IMO is less clear. The reluctance of many physicians to transplant IMO patients is based on the experience with often-complicated transplantations and a high transplant-related mortality in patients with MIOP and the chronic rather than acutely life-threatening manifestations of IMO. Here, we report 7 patients with IMO treated by HSCT after a history of recurrent osteomyelitis, debilitating osseous malformations, and pathological fractures. Patients were transplanted between ages 5 and 30 years.
All patients reported here had clinical signs and symptoms described in patients with MIOP, but that occurred here at later age. In all patients, mutations were detected in genes (TCIRG1, CA2, CLCN7, SNX10, RANK) known to cause MIOP,12,13 but these patients took much longer to acquire a transplantable disease burden than has been classically described. This is especially true of P1, who had a mutation in the SNX10 gene and who presented much later than his MIOP family members with the same mutation. Although the reason for this later presentation is currently unknown, next-generation sequencing is extending the clinical spectrum of osteopetrosis-causing genes and the associated treatment options.
P4, to the best of our knowledge, is the oldest patient transplanted with a biallelic TCIRG1 mutation. In the international osteopetrosis registry of the European Society for Immunodeficiencies and the European Society for Blood and Marrow Transplantation, comprising data of 284 patients and 165 transplanted patients (including 4 in our cohort), the median age at HSCT is 6.8 months (range, 0.5-364; mean, 17) (A.S.S., unpublished data). There are 41 patients transplanted at an age >12 months but only 12 patients (including 4 in our cohort) transplanted beyond 60 months of age. Unfortunately, in these registries, genetic data are missing for 8 patients transplanted beyond 60 months.
Regarding the indication for HSCT, the heterogeneity of genetics and clinical presentation with “overlapping” features in patients with intermediate and infantile osteopetrosis creates a real and critical challenge to define the best treatment strategy for individual patients. For patients with MIOP, immediate HSCT is the treatment of choice, provided a neurodegenerative variant can be excluded. These patients are transfusion dependent in most cases.1 However, in patients with IMO, the indication for HSCT is clearly different and more complicated. Four patients became transfusion dependent for red blood cells later in life (P2, P3, P4, and P6), but all patients suffered from complications because of their pathological bone morphology, such as pathological fractures in all 6 and destructive osteomyelitis of the maxilla or mandible in 3 patients (P2, P3, P4). These complications impair the quality of life of these patients to an extent that they and physicians have chosen the curative but high-risk treatment option of HSCT.
In patients with MIOP, transplant courses are often associated with a high risk of specific complications such as SOS/VOD, arterial pulmonary hypertension, and rejection.14,15 In our cohort of IMO patients, those transplanted with T cell–replete grafts from matched related (P2, P3, P4, P5) or MUDs (P1, P7), were engrafted and are alive. No SOS/VOD was observed. This could be partially explained by the treosulfan-based conditioning protocol for most patients. Importantly, all surviving patients demonstrated full donor chimerism with sufficient bone remodeling in the months and years following donor cell engraftment, reflecting a normalized osteoclast activity. Consequently, the clinical condition and quality of life improved markedly in all surviving patients. Although younger age is usually associated with improved transplant outcome, the youngest patient in our cohort died following 3 attempts at haploidentical transplantation. This is in line with the known observation that alternative donor transplantation is associated with an increased risk of rejection in osteopetrosis, although our cohort is too small to make such a recommendation in IMO.
With the diagnosis of IMO based on the association of clinical signs and a mutation in a gene known to cause osteopetrosis, the authors would suggest offering HSCT. As shown in these patients, the decision for a potentially life-threatening therapy can be justified because the progressive course of the disease includes osseous malformations, blood transfusions, pathological fractures, recurrent infectious osteomyelitis, loss of mobility, loss of vision, and, in summary, a considerable impairment of quality of life and most probably a reduced life expectancy.
The full-text version of the article contains a data supplement.
Acknowledgments
The authors thank the patients and their families for their faith in our care, the departmental nursing and administrative staff for their tireless commitment to patient care, and Zeev Rotstein (Director of The Hadassah Medical Center) for his support of the department and the patients.
This work was supported by the Deutsche Forschungsgemeinschaft (grant DFG WA 1597/4-1). B.S.’s position is supported by the Australian Government Research Training Program Scholarship.
Authorship
Contribution: P.S. and A.S.S. conceived and designed the study; A.S.S. and P.S. analyzed the data; P.S., S.G., B.A., I.Z., B.S., O.E., M.S., M.H., C.S., I.F., M.B., S.v.H., D.B., K.-M.D., and A.S.S. identified study subjects and collected clinical data and samples; and P.S., B.S., M.B., D.B., and A.S.S. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ansgar S. Schulz, Department of Pediatrics and Adolescent Medicine, University Medical Center Ulm, Eythstr 24, D-89075 Ulm, Germany; e-mail: ansgar.schulz@uniklinik-ulm.de.